Introduction
It has previously been shown that platelet
activation is closely associated with inflammatory bowel disease
(IBD), an autoimmune disorder that is associated with frequent
relapses (1,2). As the disease progresses, tissue
damage and microthrombi are common. Notably, mucosal capillary
thrombosis can be detected in the rectal biopsy specimens of
patients with IBD. Thrombosis mainly relies on platelet activation,
indicating a close relationship between platelet activation and IBD
(3). Furthermore, platelet
aggregation and release are commonly observed on the colonic mucosa
of patients with ulcerative colitis (UC) (4).
In the immune disease UC, chemokines such as
platelet factor 4 (PF4) are released after platelet activation in
the early stages of inflammation, recruiting innate immune cells,
such as macrophages. Macrophages serve an important role in
maintaining intestinal immune homeostasis and have a notable effect
on IBD, providing a promising opportunity for developing novel
treatments (5). Notably, an
association between platelets and macrophages has been reported in
the context of sepsis, atherosclerosis and other diseases (6-8).
Additionally, platelet-monocyte complexes (PMCs) have been shown to
indicate the severity of various diseases (9), including UC (10).
Several cytokines are released after platelet
activation, including PF4 (11).
Atherosclerosis (12), as well
as other diseases characterized by inflammation, induce the
enhanced release of PF4 from platelets. In the pathogenesis of IBD,
PF4 in plasma has also been reported to be increased (13) and to be associated with disease
activity (14). C-X-C motif
chemokine receptor 3 (CXCR3) is the receptor of PF4; PF4 recruits
macrophages and binds to CXCR3, promoting macrophages to release
proinflammatory factors. Furthermore, CXCR3 serves a notable role
in the pathogenesis of UC; CXCR3 expression has been shown to be
elevated in the peripheral blood and colon of patients with IBD
(15), and CXCR3 axis expression
is markedly higher in active IBD, indicating a role in its
pathogenesis (16). Since CXCR3
attracts a multitude of proinflammatory cells, such as neutrophils,
to the colon, inhibiting CXCR3 could be an effective means of
regulating intestinal inflammation (17). CXCR3 is a receptor found in
macrophages and other cell types (18), which has been demonstrated to
have an inflammatory effect on immune cells via the NF-κB pathway
(19) and Janus kinase/signal
transducer and activator of transcription pathway (20). Moreover, CXCR3 has been shown to
contribute to the pathogenesis of intestinal inflammation via the
MAPK pathway. By contrast, inhibition of CXCR3 has been shown to
confer protection against intestinal epithelial apoptosis (21). These previous findings suggest
that PF4/CXCR3 may serve an important role in IBD.
Notably, there is evidence that the PF4/CXCR3
pathway is responsible for recruiting neutrophils, monocytes and
lymphocytes (12,22), as well as polarizing macrophages
and inducing cell fibrosis in the vascular wall (23). However, there is still a lack of
understanding regarding the role of PF4/CXCR3 in IBD.
In the present study, the pivotal role of platelets
and macrophages in UC was investigated, and the critical role of
the PF4/CXCR3 pathway in the involvement of platelets and
macrophages was investigated. It may be hypothesized that the
current study could provide a solution to the issue of platelet
regulation of macrophages through the PF4/CXCR3 pathway, which
contributes to the progression of experimental colitis. Moreover,
the findings may provide a feasible option for the treatment of
UC.
Materials and methods
Patients
The current study was approved by the Ethics
Committee of Zhoupu Hospital (approval no. ZPYYLL-2018-02;
Shanghai, China) and was carried out in accordance with The
Declaration of Helsinki. Three healthy male donors aged 54±14 years
and three patients with UC (two male patients and one female
patient) aged 34±19 years were selected for the present study.
Peripheral colonic tissue from pediatric (age, <16 years) and
adult subjects were collected after obtaining written informed
consent from the patients or the pediatric patient's parent or
guardian. The samples were collected during colonoscopy. The
healthy donors underwent colonoscopy as part of a routine health
check-up at the hospital. The patients with UC were diagnosed by a
gastroenterologist and a pathologist. The puncture performed during
colonoscopy yielded colon tissue, which underwent hematoxylin and
eosin (H&E) staining and immunofluorescence detection using the
following antibodies: Anti-CD62P (1:200; cat. no. 60322-1-Ig;
Proteintech Group, Inc.), anti-inducible nitric oxide synthase
(iNOS; 1:200; cat. no. 18985-1-AP; Proteintech Group, Inc.),
anti-PF4 (1:100; cat. no. 21157-1-AP; Proteintech Group, Inc.) and
anti-CXCR3 (1:100; cat. no. 26756-1-AP; Proteintech Group,
Inc.).
Cell culture and treatment
THP-1 cells (cat. no. SCSP-567) were obtained from
The Cell Bank of Type Culture Collection of The Chinese Academy of
Sciences and were cultured in RPMI 1640 culture medium (cat. no.
L210KJ; Shanghai Basalmedia Technologies Co., Ltd.) supplemented
with 10% fetal bovine serum (cat. no. 098-150; Wisent, Inc.) and 1%
penicillin-streptomycin (cat. no. 15140122; Gibco; Thermo Fisher
Scientific, Inc.). The cells were maintained at 37°C with 95%
O2 and 5% CO2 in a humidified atmosphere.
Phorbol 12-myristate 13-acetate (PMA), PF4, AMG487 (CXCR3
inhibitor), PD98059 (ERK inhibitor) and SC75741 (p65 inhibitor)
were purchased from MedChemExpress (cat. nos. HY-18739, HY-P70618,
HY-15319, HY-12028 and HY-10496, respectively). Lipopolysaccharide
(LPS), which was used to induce the polarization of THP-1 cells to
a proinflammatory phenotype, was purchased from Beijing Solarbio
Science & Technology Co., Ltd. (cat. no. L8880). LPS (1
μg/ml) was added to THP-1 cells for 6 h at 37°C and the
samples were collected. PMA (100 ng/ml) was added to THP-1 cells
for 24 h at 37°C to induce the differentiation of monocytes into
macrophages (24). After adding
PMA to THP-1 cells to transform them into macrophages, the cells
were treated as required. Subsequently, the samples were treated
with AMG487 (2.5 μM) (25), PD98059 (10 μM) (26) or SC75741 (5 μM) (27) for 2 h at 37°C, after which PF4
(100 ng/ml) was added for a further 6 h at 37°C and the samples
were collected.
Platelet isolation and culture-medium
preparation
Human venous blood (5 ml) was collected from three
of the aforementioned healthy volunteers at the Zhoupu Hospital
Affiliated to Shanghai University of Medicine and Health Sciences
(Shanghai, China). A portion of the blood was placed in a sodium
citrate anticoagulant tube (BD Biosciences) and was subjected to
centrifugation at 200 × g for 15 min at room temperature, thereby
obtaining platelet-rich plasma (PRP). Prostaglandin 2 (500 ng/ml;
Sigma-Aldrich; Merck KGaA) was added to PRP to prevent platelet
activation. Platelet precipitation was carried out by
centrifugation at 800 × g for 3 min at room temperature. The
platelets were resuspended and washed in Tyrode's Salts buffer (140
mM NaCl, 10 mM NaHCO3, 2.5 mM KCl, 0.5 mM
Na2HPO4, 1 mM MgCl2, 22 mM sodium
citrate and 0.55 mM glucose; pH 6.5; Sigma-Aldrich; Merck KGaA),
counted, and added to RPMI 1640 complete culture medium in an
appropriate proportion for use (6). THP-1 cells were co-cultured with
platelets at a ratio of 1:100 for 6 h. Light-field microscopic
observations were performed and images were captured using an
inverted microscope (DMi1; Leica Biosystems).
Animals
A total of 20 healthy female Balb/c mice (age, 6-8
weeks; weight, 20-22 g) were provided by Hangzhou Qizhen
Experimental Animal Technology Co., Ltd. The experimental procedure
was conducted in accordance with the guidelines set by the National
Institutes of Health (28), and
all animal experiments were approved by the Animal Care and Use
Professional Committee of Zhoupu Hospital (approval no.
ZPYYLL-2018-02). The mice were maintained under the following
controlled conditions: Temperature, 23±1°C; humidity, 40-50%; 12-h
light/dark cycle; ad libitum access to food and water. A
7-day acclimation period was allowed for the mice prior to the
commencement of the study. The animals were euthanized if any
case-predefined humane endpoints were observed, including: i)
Weight loss, rapid loss of 15-20% of original body weight; ii)
weakness, unable to eat and drink on their own, unable to stand for
up to 24 h or unable to stand with extreme reluctance; and iii)
signs of depression and hypothermia (<37°C) without anesthesia
or sedation. No humane endpoints were met throughout the
experiments.
A murine model of dextran sulfate sodium
(DSS)-induced acute colitis
A murine model of acute colitis was established by
administering 2.5% DSS (Shanghai Yeasen Biotechnology Co., Ltd.) as
previously described (29). The
mice were randomly divided into the following four groups
(n=5/group): Control, DSS, clopidogrel and AMG487 groups.
Clopidogrel is a commonly used drug to inhibit platelet activation,
and inhibition of platelet activation can reduce the production of
platelet factors, including PF4. Following a 1-week period of
acclimation, all experimental groups with the exception of the
control group were administered a DSS solution, which was freshly
prepared and replaced every other day, and the mice freely drank
water mixed with DSS; mice were treated with AMG487 or clopidogrel
(or control) for 3 days before DSS after the 1 week acclimation
period. The clopidogrel group was administered an oral gavage of 20
mg/kg/day clopidogrel (MedChemExpress) (30,31), whereas the AMG487 group was
administered an intraperitoneal injection of 5 mg/kg/day AMG487
(32). The control and DSS model
groups were administered an intraperitoneal injection of a sterile
PBS solution containing 20% β-cyclodextrin (MedChemExpress). The
disease activity index (DAI) and histological examination (H&E
staining) were used to assess the colitis model. DAI was scored as
follows: Weight loss: 0, no weight loss; 1, 0-5%; 2, 5-10%; 3,
10-15%; 4, 15-20%. The degree of fecal softness: 0, no change; 1,
soft stool; 2, loose stools. Fecal occult blood condition: 0, the
color did not change within 2 min; 1, 1-2 min discoloration; 2,
color change within 1 min; 3, discoloration within 10 sec; 4,
immediate discoloration or hematochezia. The sum of the three
scores is the DAI score. The fecal occult blood test was performed
with the use of a fecal occult blood kit (cat. no. BA2020B; Baso
Diagnostic, Inc.). Upon completion of the animal experiments,
anesthesia was initiated with 3% isoflurane and was maintained with
1.5% isoflurane, and blood (0.5 ml) was taken from the apex of the
heart, after which, the mice were euthanized with CO2 at
a volume replacement rate of 40% vol/min according to merican
Veterinary Medical Association Guidelines for the Euthanasia of
Animals: 2020 Edition (33). The
colon was then removed for subsequent analysis. The blood samples
were allowed to stand at room temperature for 60 min prior to
centrifugation. After centrifugation at 2,000 × g for 15 min at
room temperature, serum was obtained to assess the levels of
inflammatory cytokines. The colon tissues were measured and washed
to remove non-tissue debris. Subsequently, additional colon samples
were stored at −80°C for future analysis.
Histological scoring of colon
tissues
Human and murine colon samples were sectioned after
fixation with 4% paraformaldehyde for 24 h at room temperature and
embedded in paraffin (Biosharp Life Sciences). The samples were
then cut into 5-μm sections and mounted on slides. The
sections were stained with H&E and then observed under a light
microscope. Briefly, paraffin-embedded sections were oven-dried at
60°C for 2 h, deparaffinized and stained with hematoxylin for 10
min, rinsed and stained with eosin for 30 sec, washed and sealed;
the entire procedure was operated at room temperature. Two
pathologists scored the extent of intestinal mucosal damage and
inflammatory cell infiltration in a blinded manner. Briefly, in the
case of damage to the intestinal mucosa, the following criteria
were applied: 0, no damage; 1, discrete mucosal epithelial damage;
2, superficial mucosal erosion; 3, extensive mucosal damage. For
crypt abnormalities, the following criteria were applied: 0, normal
crypts; 1, a few crypts showed structural changes or atrophy; 2,
structural changes or atrophy of numerous crypts; 3, extensive
crypt abnormalities and loss. For inflammatory cell infiltration,
the following criteria were applied: 0, none or a small amount of
inflammatory cells in the lamina propria; 1, increased numbers of
inflammatory cells in the lamina propria; 2, inflammatory cells
spread to the submucosa; 3, the whole layer has inflammatory cell
infiltration. The sum of the three scores was calculated as the
colon histological score.
Reverse transcription-quantitative PCR
(RT-qPCR)
A total of 1×106 THP-1 cells/well were
seeded into 6-well plates and divided into the following five
experimental groups: i) Control group; ii) LPS group; iii) PLT
group; iv) LPS + PLT group and v) LPS + PLT + CaCl2
group. In the PLT groups, the THP-1 cells were co-cultured with
platelets at a ratio of 1:100 for 6 h. In the LPS group, cells were
collected 6 h after stimulation with LPS. In the LPS + PLT group,
LPS was added during co-culture. In the LPS + PLT +
CaCl2 group, 10%w/v CaCl2 was added to
platelet conditioned medium for 5 min before co-culture with THP-1,
and LPS was added at the same time. CaCl2 is a commonly
used platelet activator. All experiments were performed at 37°C. In
addition, PMA (100 ng/ml) was added to THP-1 cells for 24 h at 37°C
to induce the differentiation of monocytes into macrophages; these
treatments are separate to the Control, LPS, PLT and LPS + PLT
groups. After adding PMA to THP-1 cells to transform them into
macrophages, experiments related to PF4/CXCR3 were performed.
Subsequently, the samples were treated with AMG487 (2.5
μM/ml), PD98059 (10 μM) or SC75741 (5 μM) for
2 h at 37°C, after which PF4 (100 ng/ml) (25) was added for a further 6 h at 37°C
and the samples were collected.
Total RNA was isolated from treated THP-1 cells and
mouse colon tissue samples using TRIzol® (Invitrogen;
Thermo Fisher Scientific, Inc.); briefly, the cells were washed
with PBS, the supernatant was discarded and TRIzol was added for 10
min. Subsequently, chloroform was added and mixed vigorously for 30
sec, the supernatant was left for 10 min, and was then centrifuged
at 12,000 × g for 15 min at 4°C, and an equal volume of isopropanol
was added for 10 min. After centrifugation at 12,000 × g for 15 min
at 4°C, the supernatant was discarded and then pre-cooled 75%
ethanol was added to the remaining pellet. The RNA precipitate was
obtained by centrifugation at 7,500 × g for 5 min at 4°C and was
resuspended in DEPC water. RNA at a final concentration of 1
μg was reverse transcribed into cDNA using a reverse
transcriptase kit (Vazyme Biotech Co., Ltd). After mixing the
relevant reagents of the kit, the samples were incubated at 50°C
for 15 min and 85°C for 5 sec. qPCR was conducted on a Rotor-Gene
Q-cycler (Qiagen, Inc.) using the Rotor-Gene SYBR Green qPCR Kit
(Vazyme Biotech Co., Ltd.), according to the manufacturer's
protocol. The thermocycling conditions were as follows: 95°C for 30
sec, followed by 40 cycles at 95°C for 10 sec and 60°C for 30 sec;
and final steps at 95°C for 15 sec, 60°C for 1 min and 95°C for 15
sec. Human and mouse GAPDH were used as internal controls for
parallel amplification. Relative changes in gene expression were
quantified using the 2−ΔΔCq method (34,35). All experiments were performed in
triplicate. The specific primers (Sangon Biotech Co., Ltd.) used
are listed in Table I. Among the
specific primers, SRC was found in the 'Chemokine Signaling
Pathway' and 'Cytokine-Cytokine Receptor Interaction' pathways when
searching the downstream targets of PF4/CXCR3 in the Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathways (KEGG PATHWAY:
map04062; https://www.genome.jp/entry/map04062 and KEGG PATHWAY:
map04060; https://www.genome.jp/entry/map04060).
 | Table IPrimer sequences used for
quantitative PCR. |
Table I
Primer sequences used for
quantitative PCR.
A, Human primers
|
|---|
| Gene | Forward primer,
5′-3′ | Reverse primer,
5′-3′ |
|---|
| GAPDH |
GGAGCGAGATCCCTCCAAAAT |
GGCTGTTGTCATACTTCTCTCATGG |
| IL-1β |
ATGGCTTATTACAGTGGCAATGAGG |
TGTAGTGGTGGTCGGAGATTCG |
| IL-6 |
TTCGGCAAATGTAGCATG |
AATAGTGTCCTAACGCTCATAC |
| TNF-α |
ATGAGCACTGAAAGCATGATCCG |
AGGAGAAGAGGCTGAGGAACAAG |
| IL-10 |
CTTGCTGGAGGACTTTAAGGGTTAC |
CTTGATGTCTGGGTCTTGGTTCTC |
| PF4 |
AACGGAGAGCCTGCTGAGTG |
CCCAGACAGAAGTTGTTCTAACCAG |
| CXCR3 |
TGGTGGTGCTGGTGGACATC |
GCCTGAGGTGACCGACTTGG |
| ERK |
TGGTGTGCTCTGCTTATGATAATGTC |
AGTAGGTCTGGTGCTCAAAGGG |
| P65 |
CCTGTCCTTTCTCATCCCATCTTTG |
GCTGCCAGAGTTTCGGTTCAC |
| SRC |
TCCAAGCCGCAGACTCAGG |
CATCCACACCTCGCCAAAGC |
|
| B, Mouse
primers |
|
| Gene | Forward primer,
5′-3′ | Reverse primer,
5′-3′ |
|
| GAPDH |
AAGAAGGTGGTGAAGCAGG |
GAAGGTGGAAGAGTGGGAGT |
| IL-1β |
GAAATGCCACCTTTTGACAGTG |
TGGATGCTCTCATCAGGACAG |
| IL-6 |
TAGTCCTTCCTACCCCAATTTCC |
TTGGTCCTTAGCCACTCCTTC |
| TNF-α |
CCGAGATGTGGAACTGGCAGAG |
CCACGAGCAGGAATGAGAAGAGG |
| ZO-1 |
AGAGCAAGCCTTCTGCACAT |
TCGGGTTTTCCCTTTGAAGAGT |
| OCLN |
ACATGTATGGCGGAGAGATGC |
GGGGCGACGTCCATTTGTAG |
| MUC-2 |
GAAGCCAGATCCCGAAACCA |
GAATCGGTAGACATCGCCGT |
Western blotting
Total protein was isolated from THP-1 cells using
RIPA buffer (Biosharp Life Sciences) containing protease and
phosphatase inhibitors (Biosharp Life Sciences) on ice for 30 min.
Subsequently, the cell fragments were centrifuged at 12,000 × g for
15 min at 4°C. The soluble protein component was mixed with 5X
loading buffer (Biosharp Life Sciences), and the concentration of
proteins was determined using the BCA protein assay kit (Beyotime
Institute of Biotechnology). Equal amounts of protein (30
μg/lane) were subjected to SDS-PAGE on 10 or 12.5% gels, and
were electroblotted onto PVDF membranes (MilliporeSigma). The
membranes were blocked using 5% fat-free milk (Beyotime Institute
of Biotechnology) for 2 h at room temperature and were then
incubated at 4°C overnight with the following primary antibodies:
Anti-CD62P (1:2,000; cat. no. 60322-1-Ig; Proteintech Group, Inc.),
anti-inducible nitric oxide synthase (iNOS; 1:500; cat. no.
18985-1-AP; Proteintech Group, Inc.), anti-CD86 (1:500; cat. no.
26903-1-AP; Proteintech Group, Inc.), anti-β-actin (1:20,000; cat.
no. 66009-1-Ig; Proteintech Group, Inc.), anti-Bcl-2 (1:500; cat.
no. 66799-1-Ig; Proteintech Group, Inc.), anti-Bax (1:1,000; cat.
no. ET1603-34; HUABIO), anti-caspase-3 (1:1,000; cat. no. 14220;
Cell Signaling Technology, Inc.), anti-ERK1/2 (1:1,000; cat. no.
4695; Cell Signaling Technology, Inc.), anti-phosphorylated
(p)-ERK1/2 (1:2,000; cat. no. 4370; Cell Signaling Technology,
Inc.), anti-p65 (1:1,000; cat. no. 6956; Cell Signaling Technology,
Inc.) and anti-p-p65 (1:1,000; cat. no. 3033; Cell Signaling
Technology, Inc.). After incubation with horseradish
peroxidase-conjugated secondary antibodies (1:5,000; cat. nos.
SA00001-1 and SA00001-2; Proteintech Group, Inc.) for 1 h at room
temperature, the membranes were incubated with enhanced
chemiluminescence (ECL) FemtoLight Substrate (cat. no. SQ201L;
Epizyme; Ipsen Pharma). Protein detection was performed using the
automatic chemiluminescence image analysis system (Tanon 5200
Multi; Tanon Science and Technology Co., Ltd.). Protein expression
was semi-quantified using ImageJ (version 1.5.3; National
Institutes of Health).
Flow cytometry
Apoptosis was detected by flow cytometry. After the
cells were treated with 2.5 μM AMG487 for 2 h, 100 ng/ml PF4
was added for 24 h and the harvested cells were washed three times
with PBS. Apoptosis was detected by flow cytometry after staining
with Annexin V-FITC and PI (Beyotime Institute of Biotechnology)
for 30 min at room temperature. The percentage of apoptotic cells
was calculated, and the data were analyzed using Novo Express
Immunofluorescence Analysis(version 1.4.1; Agilent Technologies,
Inc.). The apoptotic rate was determined using a NovoCyte 2000 Flow
Cytometer (Agilent Technologies, Inc.) (36).
Measurement of inflammatory
cytokines
Mouse serum IL-1β and TNF-α levels were measured
using an Automatic Biochemistry Analyzer (XPT; Siemens AG).
Reactive oxygen species (ROS)
detection
ROS levels in THP-1 cells treated with PF4 and
AMG487 were measured according to the instructions of the ROS kit
(Beyotime Institute of Biotechnology). PMA (100 ng/ml) was added to
THP-1 cells for 24 h at 37°C to induce the differentiation of
monocytes into macrophages. Subsequently, the samples were treated
with AMG487 (2.5 μM) for 2 h at 37°C, after which PF4 (100
ng/ml) was added for a further 24 h at 37°C and the samples were
collected. After washing with PBS, proportionally diluted DCFH-DA
probes (in serum-free medium at a ratio of 1:1,000) were added to
the cells. THP-1 cells were washed three times with PBS to
completely remove the unbound DCFH-DA probe and were then fixed
with 4% paraformaldehyde for 10 min at room temperature. After
nuclear staining with DAPI, the intracellular ROS levels were
assessed using ImageJ (version 1.5.3; National Institutes of
Health) by detecting the fluorescence brightness of the DCFH-DA
probe under FITC parameters. Imaging was performed using an
inverted fluorescence microscope (DMi8; Leica Biosystems).
Mitochondrial membrane potential
detection
The levels of JC-1 in THP-1 cells were determined in
accordance with the instructions provided in the JC-1 kit (Beyotime
Institute of Biotechnology). PMA (100 ng/ml) was added to THP-1
cells for 24 h at 37°C to induce the differentiation of monocytes
into macrophages. Subsequently, the samples were treated with
AMG487 (2.5 μM) for 2 h at 37°C, after which PF4 (100 ng/ml)
was added for a further 6 h at 37°C and the samples were collected.
After washing with PBS, the JC-1 dye was added to the cells at a
1:200 dilution in JC-1 staining buffer and incubated at 37°C for 20
min. Following two washes of the THP-1 cells with JC-1 staining
buffer to ensure the complete elimination of unbound JC-1 dye, the
intracellular mitochondrial membrane potential levels were
evaluated using a fluorescence microscope. This involved the
detection of the fluorescence intensities of both red and green
fluorescence. Imaging was performed using an inverted fluorescence
microscope (DMi8; Leica Biosystems).
Immunofluorescence staining analysis
For human and mouse colon tissue sample sections,
immunostaining was performed using standard procedures. Briefly,
human and murine colon samples were sectioned after fixation with
4% paraformaldehyde for 24 h at room temperature and embedded in
paraffin (Biosharp Life Sciences). Paraffin-embedded 5-μm
sections were dewaxed using xylene and rehydrated using gradient
alcohol, and antigen repair was performed by microwave heating with
Tris-EDTA (pH 9.0) at 95°C for 20 min. The tissue was then blocked
with 5% bovine serum albumin (cat. no. ST023; Beyotime Institute of
Biotechnology) for 30 min at room temperature, after which the
solution was removed and the sections were incubated overnight at
4°C with the corresponding primary antibodies. The sections were
then stained with TSA fluorescent labeling reagent (cat. no.
bry-0023-100; Shanghai Ruiyu Biotechnology Co., Ltd.) for 30 min at
room temperature and DAPI working solution (cat. no. P0131;
Beyotime Institute of Biotechnology) was added dropwise. Imaging
was carried out using a laser scanning confocal microscope (TCS
SP8; Leica Biosystems). The antibodies used in this experiment
included: Anti-CD62P (1:200; cat. no. 60322-1-Ig; Proteintech
Group, Inc.), iNOS (1:100; cat. no. 18985-1-AP; Proteintech Group,
Inc.), CD206 (1:200; cat. no. 18704-1-AP; Proteintech Group, Inc.),
PF4 (1:100; cat. no. 21157-1-AP; Proteintech Group, Inc.) and CXCR3
(1:100; cat. no. 26756-1-AP; Proteintech Group, Inc.).
TUNEL assay
Briefly, human and murine colon samples were
sectioned after fixation with 4% paraformaldehyde for 24 h at room
temperature and embedded in paraffin. Paraffin-embedded mouse colon
sections (5 μm) were dewaxed using xylene and rehydrated
using gradient alcohol. Sample permeabilization was performed
through incubation with 20 μg/ml proteinase K for 20 min at
room temperature. Endogenous peroxidase was inactivated via
incubation with 3% H2O2 for 5 min at room
temperature. The TUNEL reaction mixture (cat. no.
Roche-11684795910; Sigma-Aldrich; Merck KGaA) was added to the
sample and incubated for 1 h at 37°C. The DAPI working solution was
added dropwise and imaging was carried out using a laser scanning
confocal microscope (TCS SP8; Leica Biosystems).
Statistical analysis
Data are presented as the mean ± standard deviation
(n≥3). Statistical analysis was performed using either unpaired,
two-tailed Student's t-test, or one-way ANOVA followed by
Bonferroni post hoc test using GraphPad Prism (version 9.0;
Dotmatics). Categorical data, such as DAI and pathological scores
in animal experiments, are presented as the median (range) and were
analyzed using the non-parametric Kruskal-Wallis test followed by
Dunn's post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Platelets interact with monocytes and
macrophages in UC, thereby facilitating the transformation of
monocytes into macrophages that assume a proinflammatory role
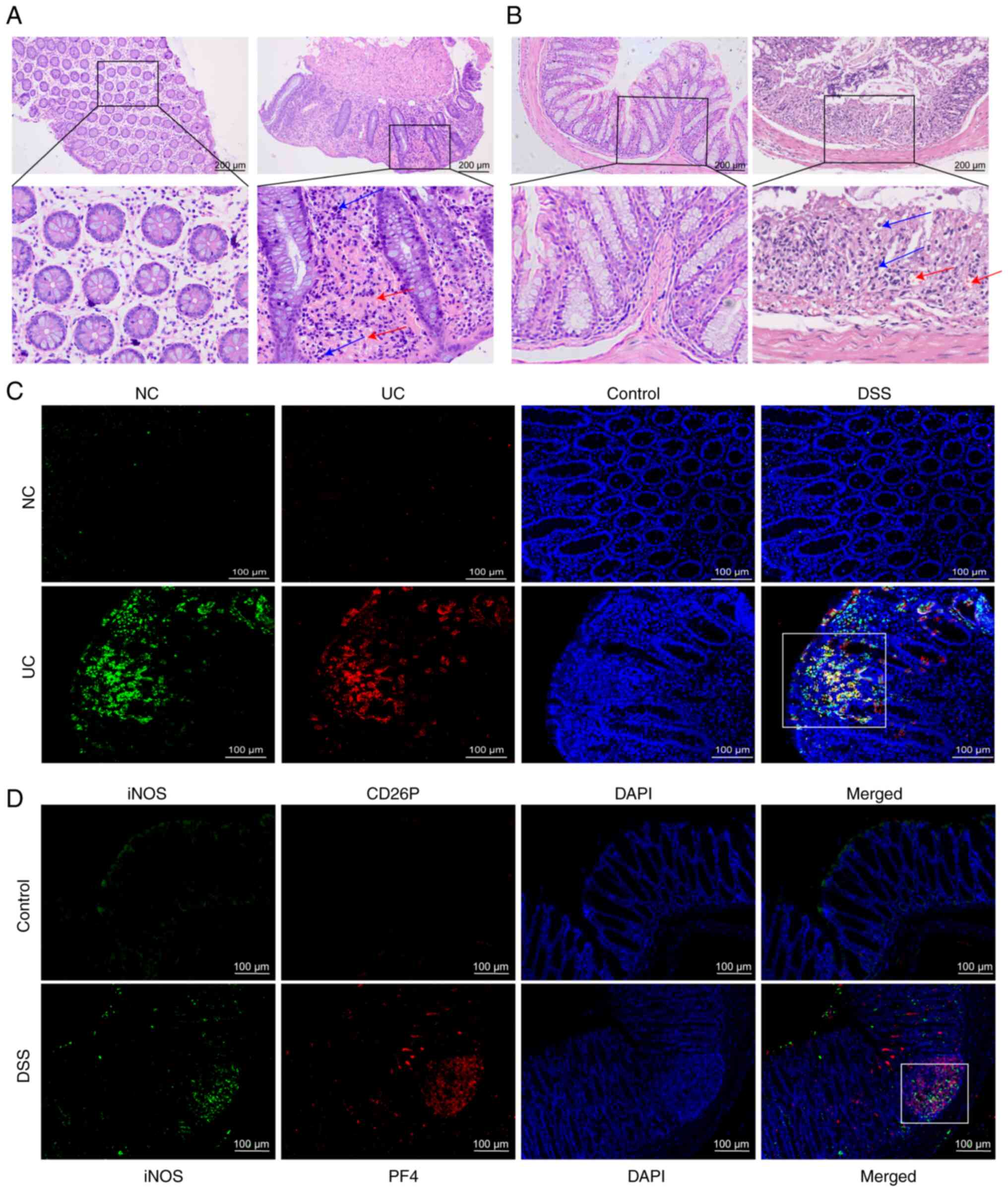
In present study, it was observed that, compared
with in the normal control group, in the colon of patients with UC,
bleeding at the colonic mucosa was shown to occur during active UC
(Fig. 1A). This was also
observed in the colon of DSS-induced mice, with more
neovascularization and microvessels, suggesting that platelets
enter the colon from blood vessels and exert their effects
(Fig. 1A and B). The
co-localization of platelets and macrophages was investigated by
staining for a platelet activation marker (CD62P) and a macrophage
M1 marker (iNOS); the findings revealed that the co-localization of
platelets with macrophages was more pronounced in the colon of
patients with UC compared with in healthy controls (Fig. 1C). This finding was confirmed by
staining for PF4 and iNOS in the mouse colon to detect the
colocalization of platelets with proinflammatory macrophages
(Fig. 1D).
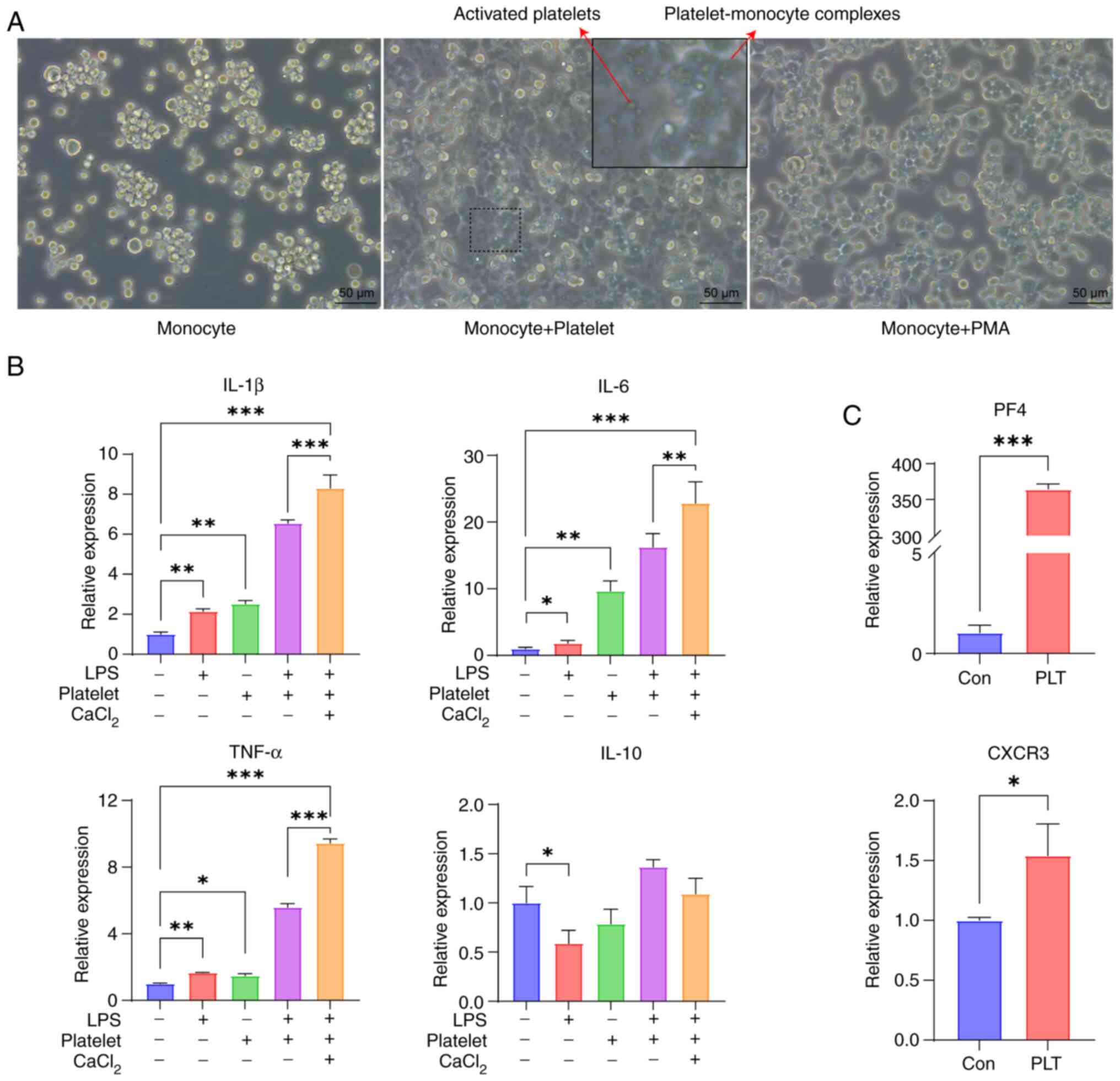
In vitro, platelets were extracted from
healthy individuals and co-cultured with THP-1 human monocytes.
Observations at the microscopic level revealed that platelets
contributed to the transformation of monocytes into macrophages,
THP-1 cells were transformed from suspended monocytes into adherent
macrophages (Fig. 2A). PMA, a
common drug that induces mononuclear-to-macrophage transformation,
was used as a reference group to verify THP-1 adherence. To examine
the effects of platelets on macrophages in an inflammatory
environment, an in vitro model of an inflammatory
environment induced by LPS was developed. RT-qPCR confirmed that
platelets facilitated the release of the proinflammatory cytokines
IL-1β, IL-6 and TNF-α from monocyte-derived macrophages.
Furthermore, in an LPS-induced proinflammatory environment, the
addition of CaCl2 to platelets resulted in a more
pronounced inflammatory response. By contrast, platelets had little
impact on the anti-inflammatory cytokine IL-10 (Fig. 2B). After co-culturing THP-1 cells
with platelets, RT-qPCR demonstrated a notable increase in the
expression of PF4 as well as CXCR3 compared with THP-1 cells only
(Fig. 2C). According to these
results, platelets may serve a key role in facilitating the
differentiation of monocytes into macrophages and exerting
proinflammatory effects.
Interaction between platelets and
macrophages occurs via the PF4/CXCR3 pathway, which facilitates the
polarization of macrophages towards the M1 phenotype
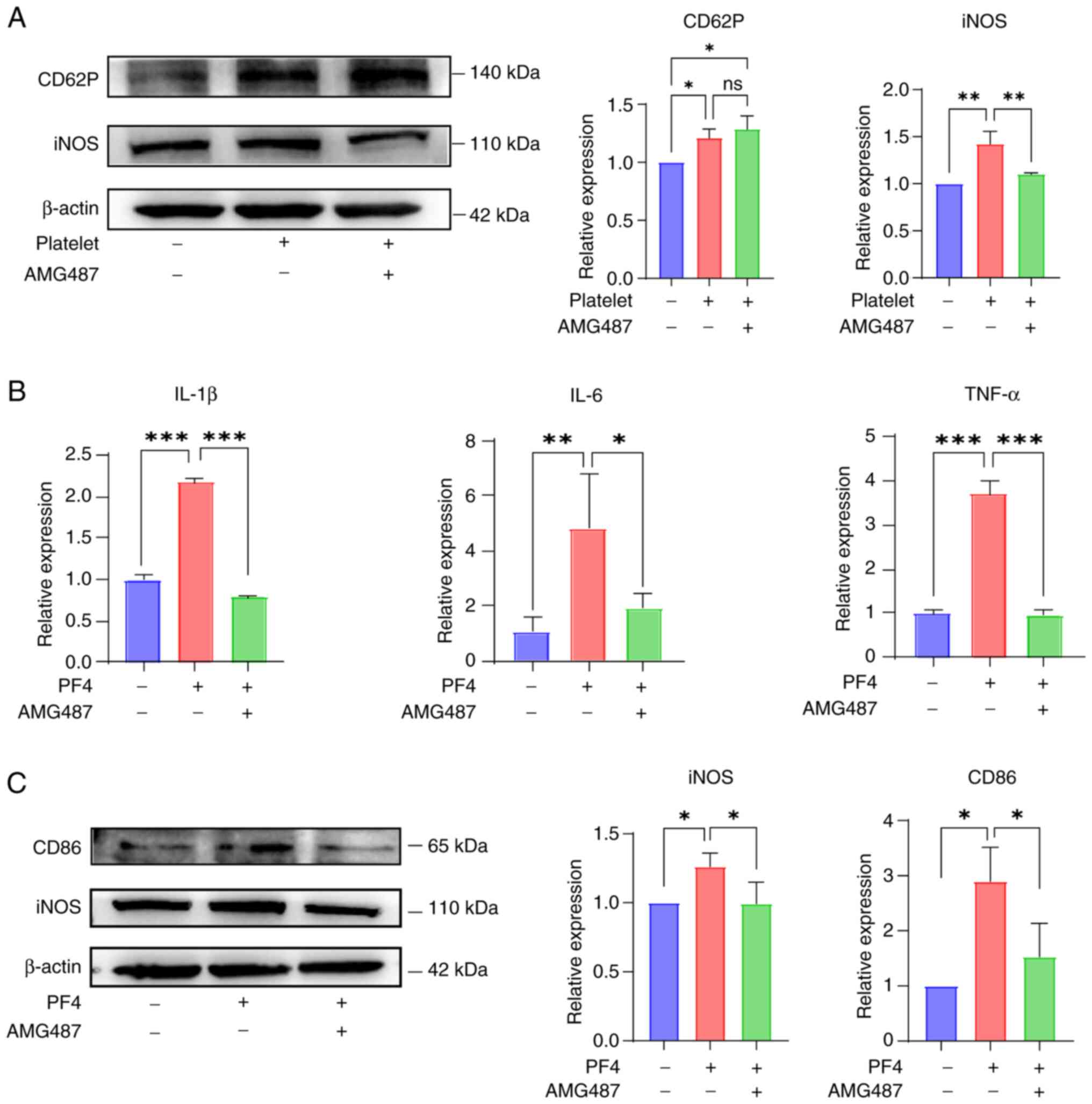
It was hypothesized that platelet activation could
influence macrophages through the PF4/CXCR3 pathway, thereby
affecting the progression of UC. Therefore, the CXCR3 inhibitor
AMG487 was used during co-culture to validate its efficacy. As a
result of the co-culture, the expression levels of CD62P, a
platelet activation marker, and iNOS, a macrophage M1-type marker,
were increased. In the presence of AMG487, iNOS expression was
reduced, whereas the expression of CD62P was not affected (Fig. 3A). To better understand the role
of this pathway, PF4 protein was added to THP-1 cells and the
levels of inflammatory markers were measured. The results of
RT-qPCR demonstrated that, as per the observations shown in
Fig. 2B, the expression levels
of inflammatory indicators, such as IL-1β, IL-6 and TNF-α, were
elevated after the addition of PF4, whereas these expression levels
were reduced following the addition of AMG487 (Fig. 3B).
According to the aforementioned findings, it was
indicated that the addition of PF4 enhanced the M1 proinflammatory
phenotype, as demonstrated by the M1 phenotypic markers CD86 and
iNOS. Both CD86 and iNOS were upregulated after the addition of PF4
to THP-1 cells, whereas the inhibition of CXCR3 suppressed
inflammation in macrophages (Fig.
3C). These results suggested that platelets interact with
macrophages through the PF4/CXCR3 pathway, thereby promoting
macrophage polarization towards a proinflammatory state.
PF4/CXCR3 is involved in the inflammatory
response, and affects the oxidative stress and apoptosis of
macrophages
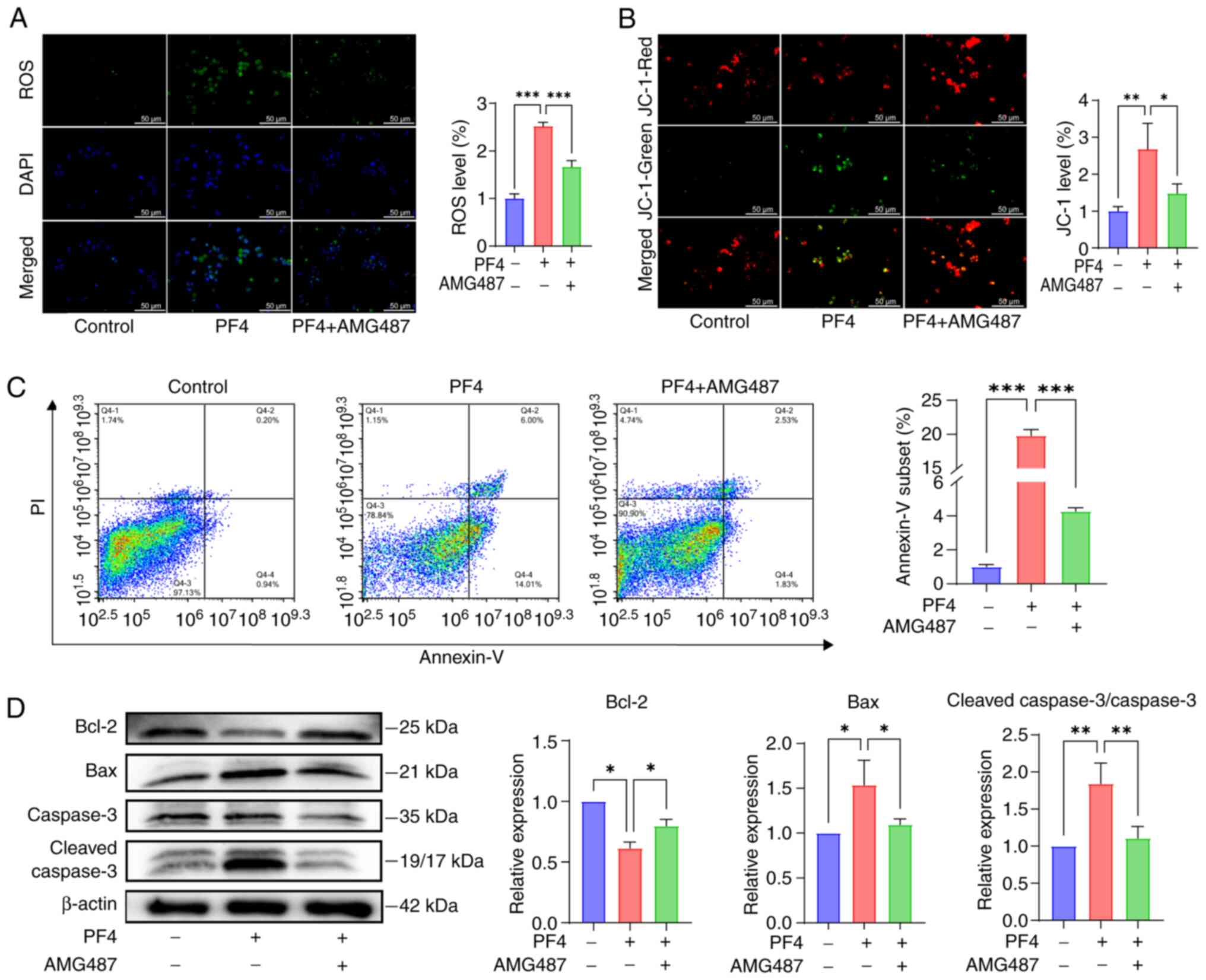
ROS have been demonstrated to be associated with the
development of inflammation. The levels of ROS were significantly
elevated following the addition of PF4, whereas they were decreased
following the addition of a CXCR3 inhibitor (Fig. 4A). Notably, the function of
mitochondria can be impaired by excessive ROS; therefore, changes
in the mitochondrial membrane potential were observed in
macrophages following the addition of PF4 by JC-1 staining. When
PF4 was added, the green fluorescence of the monomer was markedly
increased, indicating a decrease in mitochondrial membrane
potential, which AMG487 reversed (Fig. 4B). Reduced mitochondrial membrane
potential can initiate the apoptosis signaling pathway; therefore,
apoptosis was detected by Annexin-V FITC. FITC can be used to
detect cells entering early apoptosis, PI detects cell necrosis,
whereas cells positively stained for both FITC and PI are cells
entering late apoptosis. FITC/PI staining was significantly
increased following the addition of PF4, whereas this was reversed
after the addition of AMG487 (Fig.
4C). To further clarify the regulatory factors of apoptosis,
the key proteins involved in this process were detected. Compared
with in the control group, the PF4 group exhibited significantly
higher expression levels of the pro-apoptotic protein Bax, and
lower expression levels of the anti-apoptotic protein Bcl-2,
suggesting that PF4 increased apoptosis. In addition, caspase-3,
which is a marker of irreversible apoptosis, was cleaved in
response to PF4, whereas AMG487 reversed this effect (Fig. 4D).
PF4/CXCR3 pathway mediates the
proinflammatory effect of macrophages through the MAPK and NF-κB
pathways
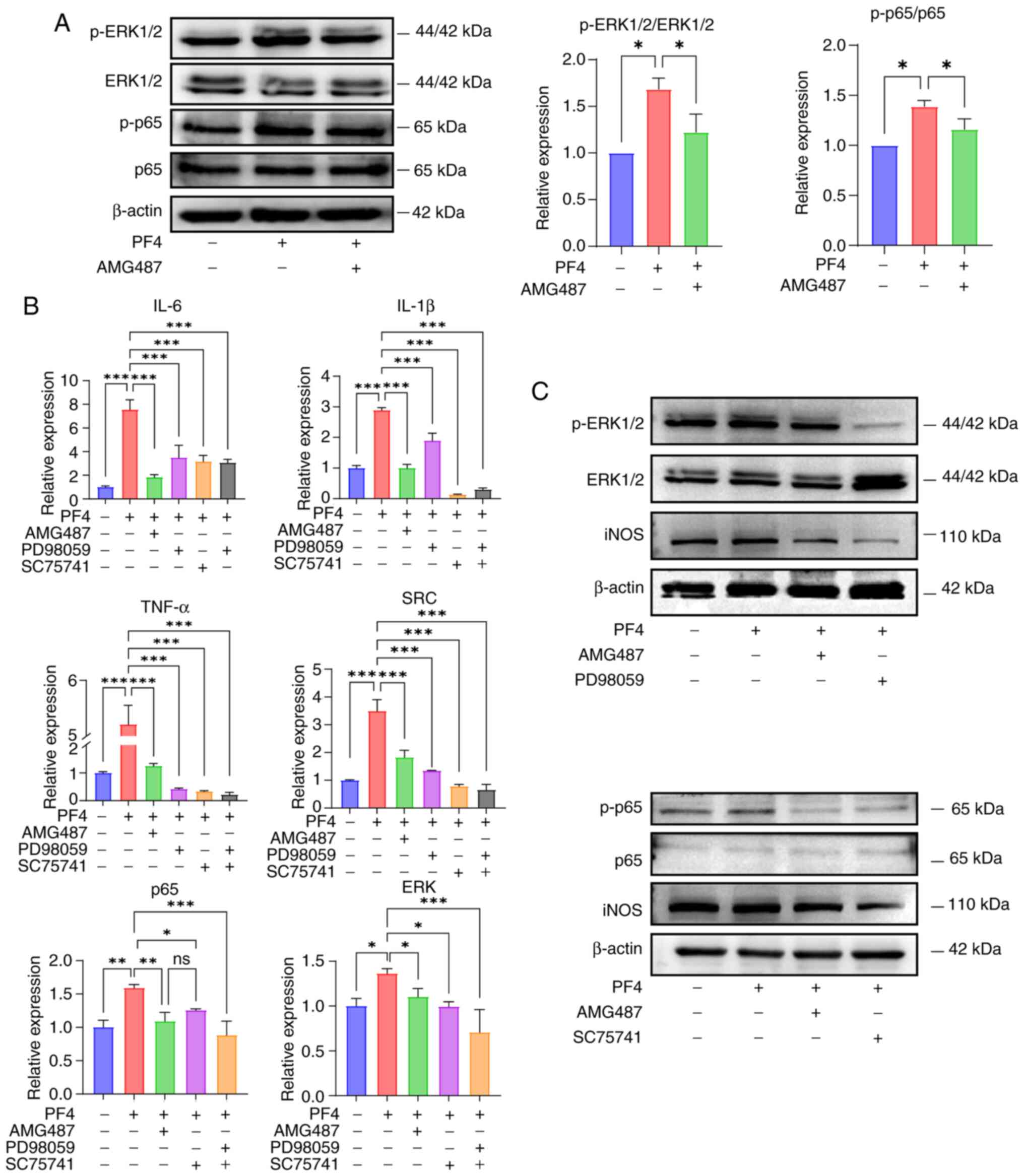
The current study examined the regulatory function
of PF4/CXCR3 by focusing on two established pathways associated
with inflammatory processes: i) NF-κB and ii) MAPK. The protein
expression levels of p-p65 and p-ERK were increased in response to
PF4 and were decreased when CXCR3 was inhibited (Fig. 5A). To confirm this finding, the
ERK inhibitor PD98059 and the p65 inhibitor SC75741 were used to
treat the cells, and the inflammatory factors and macrophage M1
marker iNOS were subsequently detected, as well as another key gene
downstream of CXCR3 and upstream of the two pathways, SRC. In the
KEGG pathways 'Chemokine Signaling Pathway' and 'Cytokine-Cytokine
Receptor Interaction', PF4 acts as a chemokine and activates
downstream signals by binding to CXCR3. SRC may be involved in
signal amplification and regulation downstream of CXCR3, and affect
the activation and migration of immune cells. The results showed
that the expression levels of iNOS, the M1 proinflammatory
phenotypic marker, and ERK, p65 and SRC, the key genes of the two
pathways, were decreased after the addition of the inhibitors, and
the effect was equally efficacious when the two inhibitors were
added together (Fig. 5B and C).
Therefore, it was hypothesized that PF4/CXCR3 may regulate
macrophage inflammation through the NF-κB and MAPK pathways.
Inhibition of platelet activation or
CXCR3 improves disease progression in a DSS-induced mouse model of
UC
The mice were treated with clopidogrel or AMG487 for
3 days prior to UC induction, followed by 7 days of free drinking
of 2.5% DSS (Fig. 6A). During
DSS administration, body weight was measured and fecal occult blood
testing was performed daily, and the DAI score was calculated based
on body weight change, fecal softness and occult blood conditions.
Following the addition of DSS, the weight of mice decreased
significantly, whereas the DAI score was significantly increased.
Both the body weight and DAI of mice were significantly improved
after administration of clopidogrel or AMG487 (Fig. 6B and C). The length of the colon
is also a factor in determining UC. In the DSS group, the colon was
significantly shortened, which was reversed in the clopidogrel and
AMG487 groups (Fig. 6D). As a
result of H&E staining, colon pathology was assessed and scored
according to relevant inflammatory factors. The results indicated
that the DSS group had the highest pathological score and the most
severe disease, whereas clopidogrel and AMG487 effectively
alleviated the condition (Fig.
6E).
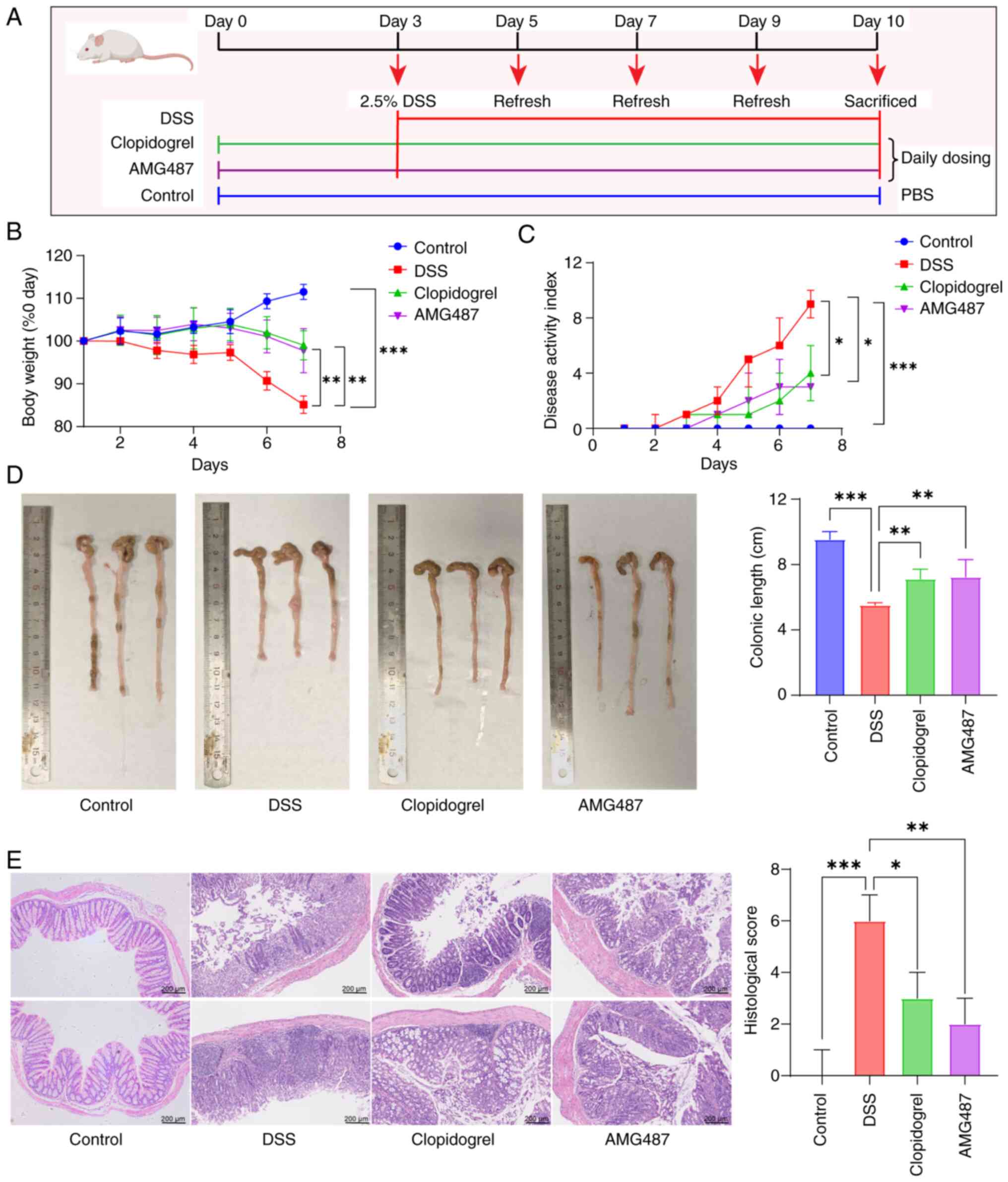 | Figure 6Platelet or C-X-C motif chemokine
receptor 3 inhibition may prevent UC in mice. (A) Flow chart for
modeling of UC in Balb/c mice; after 3 days of pretreatment with
clopidogrel or AMG487, mice were given 2.5% DSS solution freely.
The DSS solution was changed every 2 days and the mice were
sacrificed on the last day of modeling. (B) Body weight of mice
during DSS modeling was recorded daily. (C) Daily disease activity
index score of mice during modeling, which was calculated according
to changes in body weight, stool softness and fecal occult blood.
(D) After modeling, the colon length of mice in each group was
recorded and compared (n=5). (E) At the end of modeling, the colons
were collected and pathological sections were made, and the
pathological scores were calculated according to the degree of
inflammatory cell infiltration, degree of crypt abnormality and
degree of mucosal injury measured by hematoxylin and eosin
staining. Scale bar, 200 μm. Continuous data are presented
as the mean ± SD, whereas categorical data are presented as the
median (range) (n=5). *P<0.05, **P<0.01
and ***P<0.001. UC, ulcerative colitis; DSS, dextran
sulfate sodium. |
PF4/CXCR3 pathway aggravates the
progression of UC, and blocking the release of PF4 from platelets
or inhibiting CXCR3 can effectively inhibit UC
As a result of fluorescent double labeling of colon
macrophage M1 and M2 markers (iNOS/CD206), an increase in M1
proinflammatory expression and a decrease in M2 expression in the
mouse colon was observed after DSS stimulation, which was
ameliorated by clopidogrel and AMG487 (Fig. 7A). As demonstrated by peripheral
blood measurement of inflammatory cytokines and colon RT-qPCR,
inflammation was elevated in the DSS group, which was ameliorated
in the clopidogrel and AMG487 groups (Fig. 7B and C). Additionally, double
labeling of PF4 and CXCR3 was performed. In the DSS group, confocal
staining of PF4 and CXCR3 was detected, indicating that UC was
aggravated by PF4/CXCR3 signaling in mice. Among the treatments
administered, clopidogrel could inhibit the activation of platelets
and reduce the content of PF4, whereas AMG487 inhibited CXCR3
expression (Fig. 7D). This
finding was also confirmed by PF4/CXCR3 double labeling on colon
samples from patients with UC (Fig.
7E).
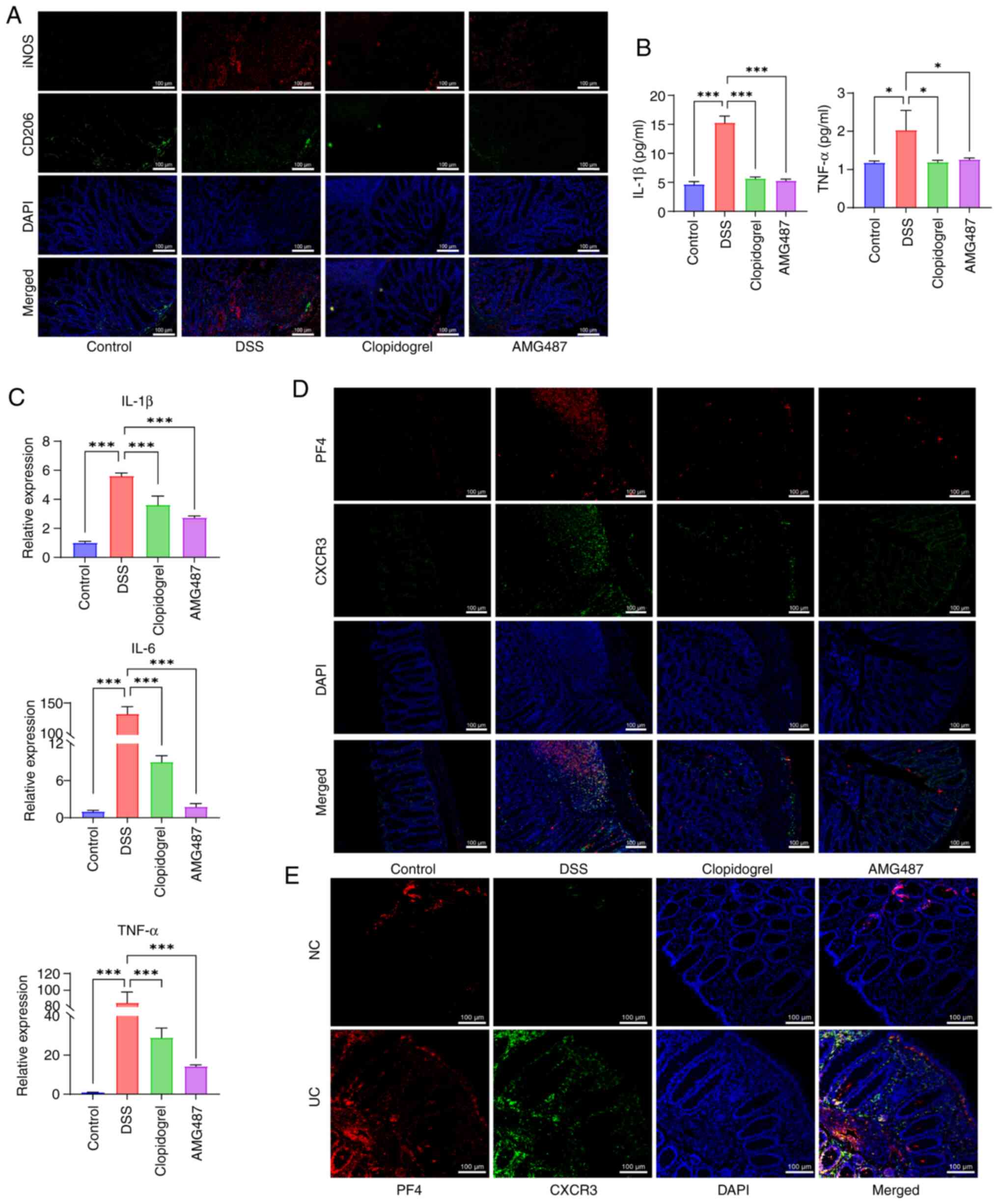 | Figure 7PF4/CXCR3 pathway affects the
development of inflammation in a mouse model of UC. (A)
Immunofluorescence visualization of M1 (iNOS)/M2 (CD206) expression
in murine macrophages. Scale bar, 100 μm. (B) Changes in the
serum levels of proinflammatory cytokines IL-1β and TNF-α in each
group of mice (n=3). (C) Expression levels of mouse proinflammatory
cytokines, IL-1β, IL-6 and TNF-α, were measured by reverse
transcription-quantitative PCR in colon tissue (n=3). (D)
Immunofluorescence detection of PF4/CXCR3 expression and
distribution in mouse colon samples. Scale bar, 100 μm. (E)
Immunofluorescence detection of PF4/CXCR3 expression and
distribution in clinical colon samples. Scale bar, 100 μm.
Data are presented as the mean ± SD (n=3). *P<0.05
and ***P<0.001. NC, normal control; UC, ulcerative
colitis; PF4, platelet factor 4; CXCR3, C-X-C motif chemokine
receptor 3; iNOS, inducible nitric oxide synthase; DSS, dextran
sulfate sodium. |
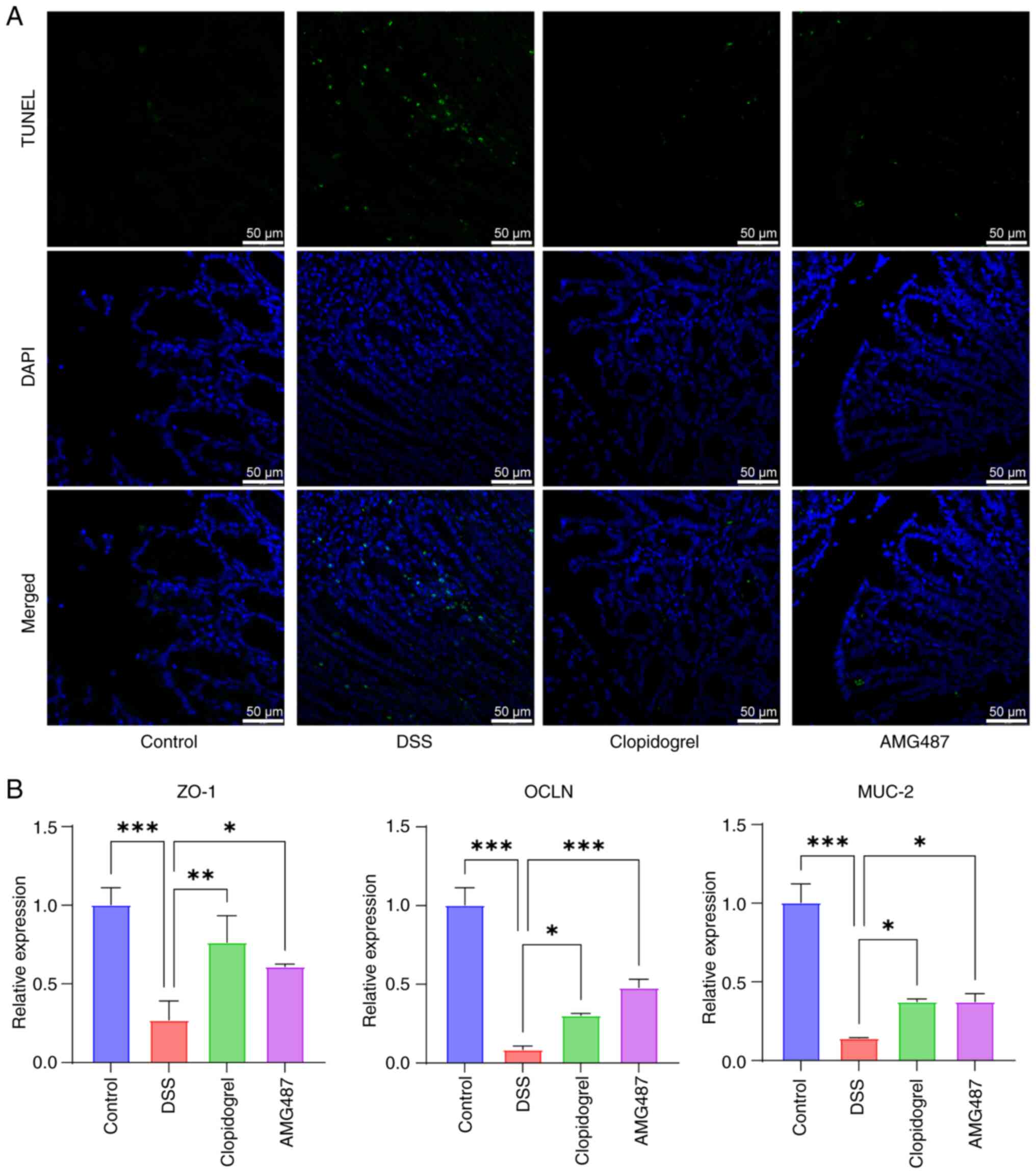
TUNEL assay was then performed to detect apoptosis
in mouse colons, and it was observed that apoptosis was markedly
enhanced in the DSS group, whereas it was reduced in the
clopidogrel and AMG487 groups (Fig.
8A). A similar pattern of results was observed when tight
junction proteins were used to measure the function of the colonic
barrier. Zonula occludens 1, mucin 2 and occludin, which serve
important roles in intestinal homeostasis, were stably expressed in
the colon of the control group. Mice in the DSS group had severe
colon tissue injury, and the expression levels of these genes were
decreased; however, clopidogrel and AMG487 could alleviate colonic
tissue damage and reverse these changes in expression levels
(Fig. 8B). Thus, it was
indicated that the PF4/CXCR3 pathway may be one of the key pathways
in the progression of UC, which not only serves a key role in
inflammatory progression, but also has a certain role in colonic
apoptosis and intestinal barrier function.
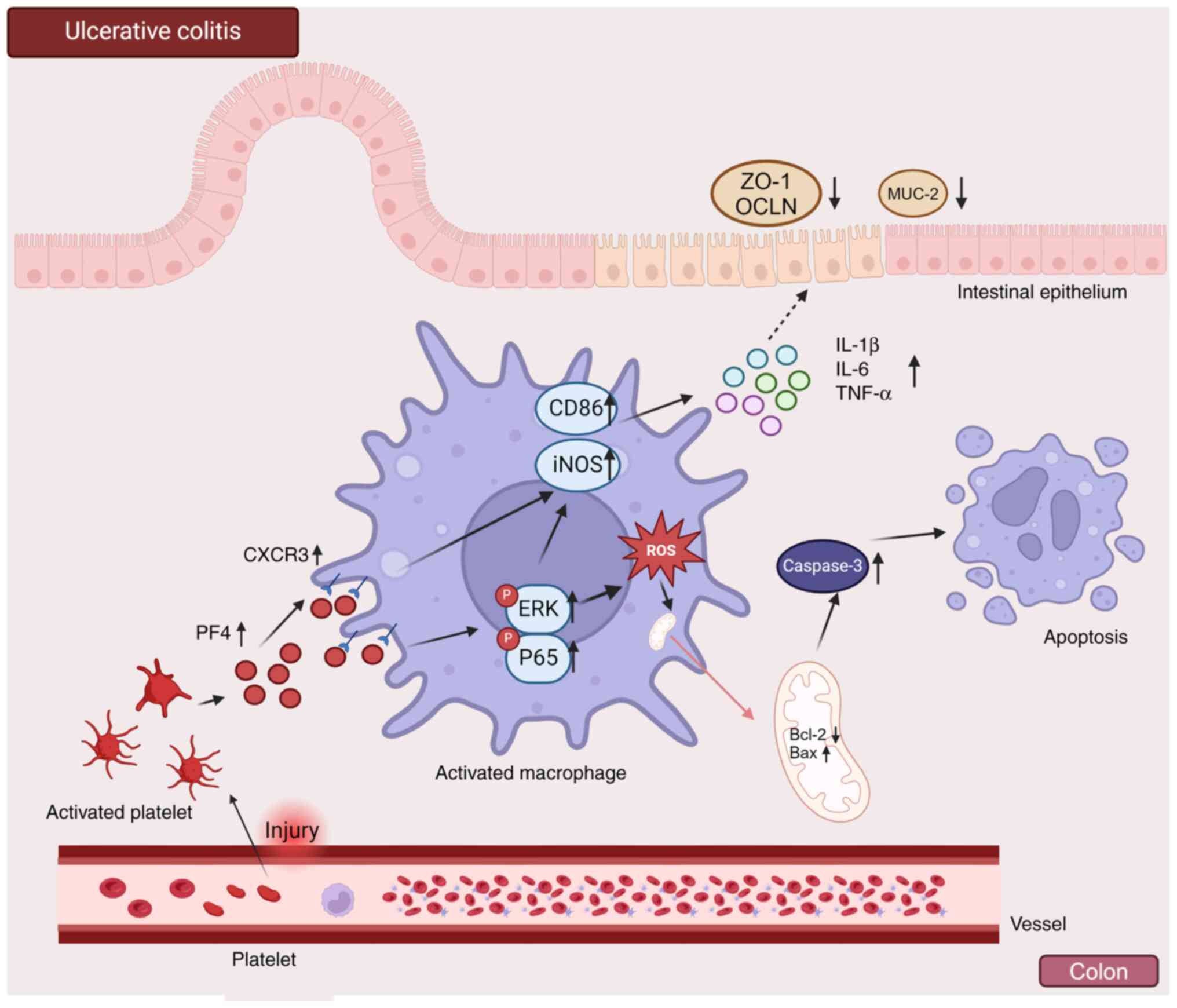
PF4/CXCR3 pathway can affect disease
progression through the NF-κB and MAPK pathways
The molecular mechanism through which platelets
regulate macrophages through PF4/CXCR3 to enhance inflammation in
UC was identified in the present study (Fig. 9). When UC develops, platelets
bleed through vascular injury and enter the colon to activate
platelets and release cytokines. PF4 released by platelets binds to
the macrophage receptor CXCR3 in the colon, activating macrophages
and polarizing them towards the proinflammatory phenotype through
signaling pathways such as the MAPK and NF-κB pathways, serving a
proinflammatory role and releasing proinflammatory factors. A
variety of proinflammatory cells may be recruited into the colon by
these inflammatory factors, causing inflammatory edema and
affecting colon cell apoptosis. Under the influence of the
PF4/CXCR3 pathway, macrophages are fully activated, ROS are
released, mitochondrial function is disrupted and macrophage
apoptosis is induced.
Discussion
IBD is an autoimmune disease that frequently recurs.
In addition, it is common for tissue damage and microthrombus
formation to occur during IBD. Platelets are associated with
thrombosis, and have the potential to adhere and accumulate at the
site of vascular injury (1). In
addition to repeated bleeding, UC is associated with common
complications such as anemia and dysplasia. Through blood vessel
bleeding, platelets enter the mucosal layer of the intestinal
mucosa. Platelets also serve a significant role in inflammatory
processes, infections and other diseases by interacting with,
stimulating and regulating innate immune cells, such as
neutrophils, monocytes and macrophages (37). It has been suggested that the
megakaryocyte/platelet lineage may have evolved to coordinate the
repair of vascular ruptures with the induction of an immune
response (38).
CD62P and PF4 are the primary markers of platelet
activation (39,40). The platelets in UC have a dual
role in hemostasis and in the inflammatory response, interacting
simultaneously with relevant immune cells in the colon. Macrophages
represent a principal cell type of the intrinsic immune response
and have been increasingly reported in UC in recent years (41). The majority of intestinal
macrophages colonize the lamina propria of the mucosa, which is
similar to the site of intestinal angiogenesis. It is reasonable to
hypothesize that platelets and macrophages interact in the gut and
stimulate each other. Once inflammation has begun, circulating
monocytes leave the bloodstream and migrate to the tissues, where
they undergo differentiation in response to growth factors,
cytokines or infection factors. This hypothesis is supported by
previous studies that have demonstrated PMC formation in other
diseases (42-45). Therefore, platelets and monocytes
were co-cultured in vitro in the present study. The
classical induction of monocyte-to-macrophage transformation with
PMA was used as a reference point. As a result of the introduction
of platelets, monocytes transitioned from suspension to apposition;
monocytes are usually maintained in suspension. After stimulation,
suspended cells can flow through blood vessels to the inflammatory
site of tissues and transform into macrophages, which colonize and
adhere to the inflammatory site. In addition, platelets attached to
monocytes and formed PMCs.
During UC, platelets enter colonic tissue and
release inflammatory factors, such as PF4 and TGF-β (4). As a result of this stimulation,
macrophages release proinflammatory cytokines, including IL-1β,
IL-6 and TNF-α (46). The
interaction between platelets and macrophages during inflammation
was simulated in vitro in the present study; notably, after
activating platelets with CaCl2, the expression levels
of proinflammatory cytokines were significantly increased, but the
expression of IL-10, an anti-inflammatory factor, was not
significantly affected. In addition, platelets may stimulate CXCR3
on mononuclear macrophages through the release of PF4 to activate
macrophages and then serve a role in inflammatory responses. In the
current study, the addition of the CXCR3 inhibitor AMG487 to the
co-culture system demonstrated that platelets may promote
inflammation by interacting with CXCR3. Despite this, platelets may
have a bidirectional immunological function. It has been
demonstrated that platelet derivatives, such as platelet lysate,
can attenuate inflammation in the late stages of inflammation
(47,48). Further investigation into these
findings is thus warranted. Taken together, the present findings
indicated that the two cell types may interact in UC, and this
interaction could contribute to the exacerbation of inflammation at
the onset of the disease.
Through PF4, platelets have an important role in a
variety of diseases (49,50).
The anticoagulant properties of PF4 are enhanced by its chemotactic
effect on neutrophils and monocytes, which contributes to the
immune response. In addition, PF4 inhibits the proliferation of
endothelial cells, suggesting that it may be involved in the
regulation of vascular processes (51). Through the release of PF4,
platelets alter the phenotype of macrophages, thereby activating
the differentiation of macrophages into proinflammatory cells; This
unique subtype of macrophages mainly triggered by PF4 are known as
M4 macrophages, which are clinically associated with inflammatory
and fibrotic processes (22,52,53). CXCR3 is one of the main targets
of PF4; notably, the CXCR3 axis serves an important role in the
progression of IBD inflammation. In addition to CXCL9, CXCL10 and
CXCL11, CXCR3 has numerous ligands that interfere with its
expression. A growing number of studies have demonstrated that
CXCL4 (PF4) binds to it as a ligand and induces targeted migration
and immune responses (54-57). Therefore, the effects of
PF4/CXCR3 on macrophages were investigated further. As a result of
the addition of PF4, macrophage inflammation was observed, which
was consistent with the results of co-culture. These findings
indicated that platelets may exert their proinflammatory effects on
macrophages by activating them via the PF4/CXCR3 pathway.
The role of PF4/CXCR3 in inflammation has also been
verified from a clinical aspect. PF4 binds to heparin to form an
antigen, produces IgG antibodies and participates in
heparin-induced thrombocytopenia. It has a variety of effects,
including interference with platelet coagulation and promotion of
host inflammatory response (58). A significant increase in PF4 has
also been observed in the serum samples of patients with IBD
(13). CXCR3 is expressed at
high levels in macrophages and other adaptive immune cells are
recruited by CXCR3. CXCR3 is expressed at high levels in Th1-Tc1
and Th17-Tc17 lymphocytes, and participate in cell migration to
inflammatory tissues (59). The
abundance of HLA-DR+CD38+ T cells, including
T regulatory cells that produce inflammatory cytokines,
CXCR3+ plasmablasts and IL1β+ macrophages and
monocytes, has been shown to be increased in colonic mucosa samples
from patients with IBD (60).
This CXCR3+ cell subpopulation serves an important role
in the pathogenesis of human IBD (15,61). Studies have also been published
on the role of the PF4/CXCR3 pathway in the context of
inflammation, cardiovascular disease and aging (49,62). Taken together, these findings
suggested that the PF4/CXCR3 signaling pathway may have an
important role in the pathogenesis of IBD.
The mechanisms that promote the progression of
inflammation were also examined in the present study. PF4/CXCR3
serves a significant role in ROS (63), apoptosis (64-66) and cell cycle progression. The
apoptosis of macrophages may be initiated by a variety of factors
during intestinal inflammation. Through a cascade of intracellular
signaling pathways, pathogen infection may induce macrophages to
initiate apoptotic programs. Additionally, various cytokines and
chemical mediators released in an inflammatory milieu may disrupt
macrophage homeostasis and trigger apoptosis. Moreover, macrophage
survival may be affected by intercellular interactions, such as
those with other immune cells (67,68). Chemokines, such as PF4, guide
immune cells to areas of inflammation. Macrophages also serve a
role in inflammation, engulfing pathogens and cytokines such as PF4
(25,49). In the present study, PF4 could
enhance the production of ROS in macrophages, disrupting the
balance between ROS production and clearance, resulting in
excessive ROS accumulation and oxidative stress, whereas inhibiting
CXCR3 decreased ROS levels. This enhanced oxidative stress induced
by PF4 may damage mitochondria (69).
To determine whether mitochondrial function was
impaired in the present study, a JC-1 assay was conducted. As a
result of adding PF4, ROS levels were increased in macrophages,
mitochondrial function was impaired and membrane potential was
decreased, which was improved by inhibiting CXCR3. Mitochondrial
membrane potential alterations are closely associated with the
initial stages of apoptosis (70). In the present study, apoptosis
was detected, and the results were consistent with those of
previous studies (64,71,72). By inhibiting the pro-apoptotic
protein Bax, Bcl-2 can protect cells from apoptosis. As a result of
reduced or inhibited Bcl-2 protein levels, the pro-apoptotic
cytoplasmic protein Bax enters mitochondria, disrupting the balance
between pro-apoptotic and anti-apoptotic proteins, resulting in
mitochondrial dysfunction and ultimately leading to macrophage
apoptosis (73). PF4 increased
ROS during inflammation, thus causing oxidative stress, which can
activate pro-apoptotic proteins. Transfer of these molecules to the
mitochondria results in a reduction in mitochondrial membrane
potential and the subsequent opening of the mitochondrial
permeability transition pore, which releases cytochrome c
(Cyt C). This release of Cyt C triggers caspase-3 (74). A caspase cascade reaction
ultimately results in a gradual increase in the externalization of
phosphatidylserine, indicating irreversible apoptosis in
macrophages (75,76).
In the present study, H&E staining of the colon
in UC models demonstrated a marked reduction in crypts, an increase
in inflammatory cell infiltration and almost complete loss of
villi, all of which could be relieved by clopidogrel or AMG487.
Pathological damage in UC may be explained by the presence of high
levels of iNOS in the crypts of the colon, which may indicate that
macrophages enter the crypts during inflammation and affect the
growth of normal cells in the colon, thereby aggravating
inflammation. Serum and colon cytokines were detected in the in
vivo model, and it was also demonstrated that platelets can
influence the polarization of macrophages and the release of
inflammatory factors by releasing cytokines, thus affecting
inflammation. It appears that PF4 and CXCR3 are clustered in areas
of severe inflammation, and that the PF4/CXCR3 pathway may
influence the severity of UC inflammation in mice.
As a result of UC, immune cells invade the crypts of
the colon, causing cryptitis and crypt loss. The release of
cytokines by platelets and macrophages during this time serves a
key role in the progression of the disease. By releasing signals,
macrophages recruit other immune cells to enter the colon and
induce apoptosis, which interferes with the ability of the colon to
repair its barrier (4,77). In murine UC, a strong association
has been detected between TUNEL-positive cells and UC severity,
indicating that apoptotic cell populations in colonic tissue may be
linked to UC severity (78). In
the present study, the TUNEL assay showed that this apoptosis was
closely related to the PF4/CXCR3 pathway, and intervention with
either of these factors could improve the development of UC. The
tight junction index of the colon was also detected in the current
study. In the mucosal epithelium, zonula occludens 1 maintains the
mechanical barrier and permeability (79), occludin maintains tight junction
stability (80) and mucin 2 is a
type of mucin secreted by goblet cells. As the main component of
intestinal mucus, mucin 2 contributes to maintenance of the
intestinal barrier and the regulation of homeostasis (81,82). The levels of these three
indicators were improved following treatment with clopidogrel and
AMG487, indicating that platelets could alter intestinal barrier
homeostasis through the PF4/CXCR3 pathway, thus aggravating UC.
There is evidence that PF4/CXCR3 modulates the
inflammation of numerous cells by affecting pathways such as NF-κB,
MAPK and others, which relates to immune dysfunction in UC
(83,84). In the present study, P65 and
ERK1/2 were detected after intervention of PF4 and CXCR3 in
vitro, and the results demonstrated that PF4/CXCR3
significantly increased the expression levels of P65 and ERK1/2,
yielding results consistent with those observed in previous
inflammation assays (85-88).
In addition, the Kyoto Encyclopedia of Genes and Genomes pathways
were searched and it was hypothesized that SRC might be a potential
target downstream of the PF4/CXCR3 pathway. As a chemokine
receptor, CXCR3 activates downstream effector molecules, including
PI3K, MAPK and SRC family kinases, through G protein-mediated
signaling (89,90). SRC serves a key role in
downstream signaling of CXCR3 and regulates cell migration and
inflammatory responses by phosphorylating downstream proteins
(11,91). Notably, SRC was expressed in the
'Chemokine Signaling Pathway' and 'Cytokine-Cytokine Receptor
Interaction' KEGG pathways. To fully demonstrate the role of these
two pathways, inhibitors of the two pathways were used to mediate
the PF4/CXCR3 pathway and the changes in inflammation were
observed. The findings further confirmed that the PF4/CXCR3 pathway
could upregulate the expression of inflammatory factors in
macrophages through the NF-κB and MAPK signaling pathways, thereby
aggravating inflammatory response and regulating apoptosis.
Notably, the present study has certain limitations.
The DSS-induced UC mouse model is an acute model, which cannot
fully mimic the long-term recurrent disease of human IBD. This
limitation can be addressed by constructing animal models of
chronic inflammation in future studies. Furthermore, the animal
model was compared with patient colon samples, and the results
showed some similarity, which will be refined in future studies. In
future studies, this limitation could be addressed by collecting
blood samples from patients to assess inflammatory factors and PF4
expression, and further studying inflammation and PF4/CXCR3
pathways in collected colon samples.
In conclusion, the THP-1 cell model induced by
platelets and PF4, and the DSS-induced IBD mouse model revealed
that platelet activation releases PF4, which may bind to macrophage
CXCR3 and activate macrophages, polarizing them towards a
proinflammatory phenotype, increasing the secretion of inflammatory
factors, stimulating oxidative stress and apoptosis, and exhibiting
proinflammatory effects. It may be possible to alleviate
inflammation by inhibiting platelet activation or CXCR3 expression.
In addition, there may be a notable role for MAPK or NF-κB
signaling pathways in PF4/CXCR3 signaling. These findings provide
an insight into the mechanisms by which PF4/CXCR3 promotes UC, and
the current study provides experimental evidence that PF4/CXCR3 may
be a promising therapeutic pathway for UC.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YZY, QP and YXN contributed to the research
conception and design. YXN and AHL performed the experiments and
drafted the manuscript. YZY, YXN and WHX confirm the authenticity
of all the raw data. WHX, RZ, RYM, LHZ and FMZ performed data
analysis and interpretation. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
The present study was performed in line with the
principles of The Declaration of Helsinki. The present study was
approved by the Ethics Committee of Shanghai Zhoupu Hospital and
the Animal Care and Use Professional Committee of Shanghai Zhoupu
Hospital (approval no. ZPYYLL-2018-02). Written informed consent
was obtained from all of the patients and the parents or guardians
of the pediatric patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Clinical Research Project
of Shanghai University of Medicine and Health Sciences (grant no.
22MC2022002), the Research Grant for Pudong Health Bureau of
Shanghai (grant no. YC-2023-0401), the Research Grant for Pudong
Health Bureau of Shanghai (grant no. PW2024D-10), the Pudong New
Area Science and Technology Commission (grant no. PKJ2023-Y109) and
the Pudong New Area Science and Technology Commission (grant no.
PKJ2021-Y32).
References
|
1
|
van der Meijden PEJ and Heemskerk JWM:
Platelet biology and functions: New concepts and clinical
perspectives. Nat Rev Cardiol. 16:166–179. 2019. View Article : Google Scholar
|
|
2
|
Dhillon AP, Anthony A, Sim R, Wakefield
AJ, Sankey EA, Hudson M, Allison MC and Pounder RE: Mucosal
capillary thrombi in rectal biopsies. Histopathology. 21:127–133.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Custodio-Chablé SJ, Lezama RA and
Reyes-Maldonado E: Platelet activation as a trigger factor for
inflammation and atherosclerosis. Cir Cir. 88:233–243.
2020.PubMed/NCBI
|
|
4
|
Huang B, Chen Z, Geng L, Wang J, Liang H,
Cao Y, Chen H, Huang W, Su M, Wang H, et al: Mucosal profiling of
pediatric-onset colitis and IBD reveals common pathogenics and
therapeutic pathways. Cell. 179:1160–1176.e1124. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pan X, Zhu Q, Pan LL and Sun J: Macrophage
immunometabolism in inflammatory bowel diseases: From pathogenesis
to therapy. Pharmacol Ther. 238:1081762022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Carestia A, Mena HA, Olexen CM, Wilczyñski
JM, Negrotto S, Errasti AE, Gómez RM, Jenne CN, Silva EA and
Schattner M: Platelets promote macrophage polarization toward
proinflammatory phenotype and increase survival of septic mice.
Cell Rep. 28:896–908.e895. 2019. View Article : Google Scholar
|
|
7
|
Laffont B, Corduan A, Rousseau M, Duchez
AC, Lee CH, Boilard E and Provost P: Platelet microparticles
reprogram macrophage gene expression and function. Thromb Haemost.
115:311–323. 2016. View Article : Google Scholar
|
|
8
|
Heffron SP, Weinstock A, Scolaro B, Chen
S, Sansbury BE, Marecki G, Rolling CC, El Bannoudi H, Barrett T,
Canary JW, et al: Platelet-conditioned media induces an
anti-inflammatory macrophage phenotype through EP4. J Thromb
Haemost. 19:562–573. 2021. View Article : Google Scholar
|
|
9
|
Rutten B, Tersteeg C, Vrijenhoek JE, van
Holten TC, Elsenberg EH, Mak-Nienhuis EM, de Borst GJ, Jukema JW,
Pijls NH, Waltenberger J, et al: Increased platelet reactivity is
associated with circulating platelet-monocyte complexes and
macrophages in human atherosclerotic plaques. PLoS One.
9:e1050192014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pamuk GE, Vural O, Turgut B, Demir M, Umit
H and Tezel A: Increased circulating platelet-neutrophil,
platelet-monocyte complexes, and platelet activation in patients
with ulcerative colitis: A comparative study. Am J Hematol.
81:753–759. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kasper B and Petersen F: Molecular
pathways of platelet factor 4/CXCL4 signaling. Eur J Cell Biol.
90:521–526. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bakogiannis C, Sachse M, Stamatelopoulos K
and Stellos K: Platelet-derived chemokines in inflammation and
atherosclerosis. Cytokine. 122:1541572019. View Article : Google Scholar
|
|
13
|
Simi M, Leardi S, Tebano MT, Castelli M,
Costantini FM and Speranza V: Raised plasma concentrations of
platelet factor 4 (PF4) in Crohn's disease. Gut. 28:336–338. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ye L, Zhang YP, Yu N, Jia YX, Wan SJ and
Wang FY: Serum platelet factor 4 is a reliable activity parameter
in adult patients with inflammatory bowel disease: A pilot study.
Medicine (Baltimore). 96:e63232017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mitsialis V, Wall S, Liu P,
Ordovas-Montanes J, Parmet T, Vukovic M, Spencer D, Field M,
McCourt C, Toothaker J, et al: Single-Cell analyses of colon and
blood reveal distinct immune cell signatures of ulcerative colitis
and Crohn's disease. Gastroenterology. 159:591–608.e510. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schroepf S, Kappler R, Brand S, Prell C,
Lohse P, Glas J, Hoster E, Helmbrecht J, Ballauff A, Berger M, et
al: Strong overexpression of CXCR3 axis components in childhood
inflammatory bowel disease. Inflamm Bowel Dis. 16:1882–1890. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chami B, Yeung AW, van Vreden C, King NJ
and Bao S: The role of CXCR3 in DSS-induced colitis. PLoS One.
9:e1016222014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pandey V, Fleming-Martinez A, Bastea L,
Doeppler HR, Eisenhauer J, Le T, Edenfield B and Storz P:
CXCL10/CXCR3 signaling contributes to an inflammatory
microenvironment and its blockade enhances progression of murine
pancreatic precancerous lesions. Elife. 10:e606462021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang C, Deng Y, Zhang Y, Ba T, Niu S,
Chen Y, Gao Y and Dai H: CXCR3 Inhibition blocks the NF-κB
signaling pathway by elevating autophagy to ameliorate
lipopolysaccharide-induced intestinal dysfunction in mice. Cells.
12:1822023. View Article : Google Scholar
|
|
20
|
Barone M, Catani L, Ricci F, Romano M,
Forte D, Auteri G, Bartoletti D, Ottaviani E, Tazzari PL, Tazzari
PL, et al: The role of circulating monocytes and JAK inhibition in
the infectious-driven inflammatory response of myelofibrosis.
Oncoimmunology. 9:17825752020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gao J, Gao J, Qian L, Wang X, Wu M, Zhang
Y, Ye H, Zhu S, Yu Y and Han W: Activation of p38-MAPK by
CXCL4/CXCR3 axis contributes to p53-dependent intestinal apoptosis
initiated by 5-fluorouracil. Cancer Biol Ther. 15:982–991.
2014.PubMed/NCBI
|
|
22
|
Domschke G and Gleissner CA: CXCL4-induced
macrophages in human atherosclerosis. Cytokine. 122:1541412019.
|
|
23
|
Hoeft K, Schaefer GJL, Kim H, Schumacher
D, Bleckwehl T, Long Q, Klinkhammer BM, Peisker F, Koch L, Nagai J,
et al: Platelet-instructed SPP1(+) macrophages drive myofibroblast
activation in fibrosis in a CXCL4-dependent manner. Cell Rep.
42:1121312023.PubMed/NCBI
|
|
24
|
Bohlen J, Zhou Q, Philippot Q, Ogishi M,
Rinchai D, Nieminen T, Seyedpour S, Parvaneh N, Rezaei N,
Yazdanpanah N, et al: Human MCTS1-dependent translation of JAK2 is
essential for IFN-γ immunity to mycobacteria. Cell.
186:5114–5134.e5127. 2023.
|
|
25
|
Ojha A, Bhasym A, Mukherjee S, Annarapu
GK, Bhakuni T, Akbar I, Seth T, Vikram NK, Vrati S, Basu A, et al:
Platelet factor 4 promotes rapid replication and propagation of
Dengue and Japanese encephalitis viruses. EBioMedicine. 39:332–347.
2019.
|
|
26
|
Hwang Y, Cha SH, Kim D and Jun HS:
Combination of PD98059 and TGF-β1 efficiently differentiates human
urine-derived stem cells into smooth muscle cells. Int J Mol Sci.
22:105322021.
|
|
27
|
Tian B, Cai D, Wang M, He T, Deng L, Wu L,
Jia R, Zhu D, Liu M, Chen S, et al: SC75741 antagonizes vesicular
stomatitis virus, duck Tembusu virus, and duck plague virus
infection in duck cells through promoting innate immune responses.
Poult Sci. 100:1010852021.PubMed/NCBI
|
|
28
|
National Research Council Committee for
the Update of the Guide for the Care and Use of Laboratory Animal:
The National academies collection: Reports funded by National
Institutes of Health. Guide for the Care and Use of Laboratory
Animals. National Academy of Sciences; Washington, DC: 2011
|
|
29
|
Zeng B, Huang Y, Chen S, Xu R, Xu L, Qiu
J, Shi F, Liu S, Zha Q, Ouyang D and He X: Dextran sodium sulfate
potentiates NLRP3 inflammasome activation by modulating the KCa3.1
potassium channel in a mouse model of colitis. Cell Mol Immunol.
19:925–943. 2022.PubMed/NCBI
|
|
30
|
Wang XL, Deng HF, Li T, Miao SY, Xiao ZH,
Liu MD, Liu K and Xiao XZ: Clopidogrel reduces
lipopolysaccharide-induced inflammation and neutrophil-platelet
aggregates in an experimental endotoxemic model. J Biochem Mol
Toxicol. 33:e222792019.
|
|
31
|
Korish AA: Clopidogrel prophylaxis abates
myocardial ischemic injury and inhibits the
hyperlipidemia-inflammation loop in hypercholestrolemic mice. Arch
Med Res. 51:515–523. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Du J, Zhang X, Han J, Man K, Zhang Y, Chu
ES, Nan Y and Yu J: Pro-Inflammatory CXCR3 impairs mitochondrial
function in experimental non-alcoholic steatohepatitis.
Theranostics. 7:4192–4203. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
American Veterinary Medical Association
(AVMA): AVMA guidelines for the euthanasia of animals: 2020
Edition. American Veterinary Medical Association; Schaumburg, IL:
2020
|
|
34
|
Yang X, Liu Z, Zhou J, Guo J, Han T, Liu
Y, Li Y, Bai Y, Xing Y, Wu J and Hu D: SPP1 promotes the
polarization of M2 macrophages through the Jak2/Stat3 signaling
pathway and accelerates the progression of idiopathic pulmonary
fibrosis. Int J Mol Med. 54:892024. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
36
|
Liu X, Li J, Huang Q, Jin M and Huang G:
Ginsenoside Rh2 shifts tumor metabolism from aerobic glycolysis to
oxidative phosphorylation through regulating the HIF1-α/PDK4 axis
in non-small cell lung cancer. Mol Med. 30:562024. View Article : Google Scholar
|
|
37
|
Mandel J, Casari M, Stepanyan M, Martyanov
A and Deppermann C: Beyond hemostasis: Platelet innate immune
interactions and thromboinflammation. Int J Mol Sci. 23:38682022.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Koupenova M, Livada AC and Morrell CN:
Platelet and megakaryocyte roles in innate and adaptive immunity.
Circ Res. 130:288–308. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cibor D, Szczeklik K, Kozioł K, Pocztar H,
Mach T and Owczarek D: Serum concentration of selected biochemical
markers of endothelial dysfunction and inflammation in patients
with the varying activity of inflammatory bowel disease. Pol Arch
Intern Med. 130:598–606. 2020.PubMed/NCBI
|
|
40
|
Takeyama H, Mizushima T, Iijima H,
Shinichiro S, Uemura M, Nishimura J, Hata T, Takemasa I, Yamamoto
H, Doki Y and Mori M: Platelet activation markers are associated
with Crohn's disease activity in patients with low C-reactive
protein. Dig Dis Sci. 60:3418–3423. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang M, Li X, Zhang Q, Yang J and Liu G:
Roles of macrophages on ulcerative colitis and colitis-associated
colorectal cancer. Front Immunol. 14:11036172023. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rolling CC, Sowa MA, Wang TT, Cornwell M,
Myndzar K, Schwartz T, El Bannoudi H, Buyon J, Barrett TJ and
Berger JS: P2Y12 inhibition suppresses proinflammatory
platelet-monocyte interactions. Thromb Haemost. 123:231–244. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sano Y, Tomiyama T, Yagi N, Ito Y, Honzawa
Y, Tahara T, Ikeura T, Fukui T, Shimoda S and Naganuma M: Platelet
activation through CD62P and the formation of platelet-monocyte
complexes are associated with the exacerbation of mucosal
inflammation in patients with ulcerative colitis. Sci Rep.
14:280552024. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chao Y, Rebetz J, Bläckberg A, Hovold G,
Sunnerhagen T, Rasmussen M, Semple JW and Shannon O: Distinct
phenotypes of platelet, monocyte, and neutrophil activation occur
during the acute and convalescent phase of COVID-19. Platelets.
32:1092–1102. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tunjungputri RN, van de Heijden W, Urbanus
RT, de Groot PG, van der Ven A and de Mast Q: Higher platelet
reactivity and platelet-monocyte complex formation in Gram-positive
sepsis compared to Gram-negative sepsis. Platelets. 28:595–601.
2017. View Article : Google Scholar
|
|
46
|
Tatiya-Aphiradee N, Chatuphonprasert W and
Jarukamjorn K: Immune response and inflammatory pathway of
ulcerative colitis. J Basic Clin Physiol Pharmacol. 30:1–10. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kim JI, Bae HC, Park HJ, Lee MC and Han
HS: Effect of storage conditions and activation on growth factor
concentration in platelet-rich plasma. J Orthop Res. 38:777–784.
2020. View Article : Google Scholar
|
|
48
|
Nebie O, Barro L, Wu YW, Knutson F, Buée
L, Devos D, Peng CW, Blum D and Burnouf T: Heat-treated human
platelet pellet lysate modulates microglia activation, favors wound
healing and promotes neuronal differentiation in vitro. Platelets.
32:226–237. 2021. View Article : Google Scholar
|
|
49
|
Schroer AB, Ventura PB, Sucharov J, Misra
R, Chui MKK, Bieri G, Horowitz AM, Smith LK, Encabo K, Tenggara I,
et al: Platelet factors attenuate inflammation and rescue cognition
in ageing. Nature. 620:1071–1079. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Liu Z, Li L, Zhang H, Pang X, Qiu Z, Xiang
Q and Cui Y: Platelet factor 4(PF4) and its multiple roles in
diseases. Blood Rev. 64:1011552024. View Article : Google Scholar
|
|
51
|
Yang C, Bachu M, Du Y, Brauner C, Yuan R,
Ah Kioon MD, Chesi G, Barrat FJ and Ivashkiv LB: CXCL4 synergizes
with TLR8 for TBK1-IRF5 activation, epigenomic remodeling and
inflammatory response in human monocytes. Nat Commun. 13:34262022.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yu B, Jia S, Chen Y, Guan R, Chen S, Tang
W, Bao T and Tian Z: CXCL4 deficiency limits M4 macrophage
infiltration and attenuates hyperoxia-induced lung injury. Mol Med.
30:2532024. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Li H, Cao Z, Wang L, Liu C, Lin H, Tang Y
and Yao P: Macrophage subsets and death are responsible for
atherosclerotic plaque formation. Front Immunol. 13:8437122022.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wang K, Wu J, Yang Z, Zheng B, Shen S,
Wang RR, Zhang Y, Wang HY, Chen L and Qiu X: Hyperactivation of
β-catenin signal in hepatocellular carcinoma recruits
myeloid-derived suppressor cells through PF4-CXCR3 axis. Cancer
Lett. 586:2166902024. View Article : Google Scholar
|
|
55
|
Kuratani A, Okamoto M, Kishida K, Okuzaki
D, Sasai M, Sakaguchi S, Arase H and Yamamoto M: Platelet factor
4-induced T(H)1-T(reg) polarization suppresses antitumor immunity.
Science. 386:eadn86082024. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Tan S, Li S, Min Y, Gisterå A, Moruzzi N,
Zhang J, Sun Y, Andersson J, Malmström RE, Wang M, et al: Platelet
factor 4 enhances CD4(+) T effector memory cell responses via
Akt-PGC1α-TFAM signaling-mediated mitochondrial biogenesis. J
Thromb Haemost. 18:2685–2700. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Tan S, Zhang J, Sun Y, Gisterå A, Sheng Z,
Malmström RE, Hou M, Peng J, Ma C, Liao W and Li N: Platelets
enhance CD4+ central memory T cell responses via platelet factor
4-dependent mitochondrial biogenesis and cell proliferation.
Platelets. 33:360–370. 2022. View Article : Google Scholar
|
|
58
|
Arepally GM: Heparin-induced
thrombocytopenia. Blood. 129:2864–2872. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Alfaro R, Llorente S, Gonzalez-Martínez G,
Jimenez-Coll V, Martínez-Banaclocha H, Galián JA, Botella C,
Moya-Quiles MR, de la Peña-Moral J, Minguela A, et al: Clinical
significance of the pre-transplant CXCR3 and CCR6 expression on T
cells in kidney graft recipients. Transplant Proc. 55:66–71. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wang S, Zhang Y, Chen G, Zhao P, Wang X,
Xu B and Yuan L: Expressions of CXCR3 and PD-1 on T cells and their
clinical relevance in colorectal cancer. Int Immunopharmacol.
132:1119882024. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Papadakis KA, Prehn J, Zhu D, Landers C,
Gaiennie J, Fleshner PR and Targan SR: Expression and regulation of
the chemokine receptor CXCR3 on lymphocytes from normal and
inflammatory bowel disease mucosa. Inflamm Bowel Dis. 10:778–788.
2004. View Article : Google Scholar
|
|
62
|
Altara R, Manca M, Brandão RD, Zeidan A,
Booz GW and Zouein FA: Emerging importance of chemokine receptor
CXCR3 and its ligands in cardiovascular diseases. Clin Sci (Lond).
130:463–478. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zhou H, Deng M, Liu Y, Yang C, Hoffman R,
Zhou J, Loughran PA, Scott MJ, Neal MD and Billiar TR: Platelet
HMGB1 is required for efficient bacterial clearance in
intra-abdominal bacterial sepsis in mice. Blood Adv. 2:638–648.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Woller G, Brandt E, Mittelstädt J,
Rybakowski C and Petersen F: Platelet factor 4/CXCL4-stimulated
human monocytes induce apoptosis in endothelial cells by the
release of oxygen radicals. J Leukoc Biol. 83:936–945. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Pervushina O, Scheuerer B, Reiling N,
Behnke L, Schröder JM, Kasper B, Brandt E, Bulfone-Paus S and
Petersen F: Platelet factor 4/CXCL4 induces phagocytosis and the
generation of reactive oxygen metabolites in mononuclear phagocytes
independently of Gi protein activation or intracellular calcium
transients. J Immunol. 173:2060–2067. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Saahene RO, Wang J, Wang ML, Agbo E and
Pang D: The antitumor mechanism of paeonol on CXCL4/CXCR3-B signals
in breast cancer through induction of tumor cell apoptosis. Cancer
Biother Radiopharm. 33:233–240. 2018.PubMed/NCBI
|
|
67
|
Liu X, Zhou M, Dai Z, Luo S, Shi Y, He Z
and Chen Y: Salidroside alleviates ulcerative colitis via
inhibiting macrophage pyroptosis and repairing the
dysbacteriosis-associated Th17/Treg imbalance. Phytother Res.
37:367–382. 2023. View Article : Google Scholar
|
|
68
|
Pedersen J, LaCasse EC, Seidelin JB,
Coskun M and Nielsen OH: Inhibitors of apoptosis (IAPs) regulate
intestinal immunity and inflammatory bowel disease (IBD)
inflammation. Trends Mol Med. 20:652–665. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Deng Z, Liu Q, Wang M, Wei HK and Peng J:
GPA Peptide-Induced Nur77 localization at mitochondria inhibits
inflammation and oxidative stress through activating autophagy in
the intestine. Oxid Med Cell Longev. 2020:49642022020. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Xiao JJ, Liu Q, Li Y, Peng FF, Wang S,
Zhang Z, Liu H, Yu H, Tao S and Zhang BF: Regulator of calcineurin
1 deletion attenuates mitochondrial dysfunction and apoptosis in
acute kidney injury through JNK/Mff signaling pathway. Cell Death
Dis. 13:7742022. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Xu W, Ye S, Liu W, Guo H, Zhang L, Wei S,
Anwaier A, Chang K, Malafaia G, Zhang H, et al: Single-cell RNA-seq
analysis decodes the kidney microenvironment induced by polystyrene
microplastics in mice receiving a high-fat diet. J
Nanobiotechnology. 22:132024. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Nevzorova TA, Mordakhanova ER, Daminova
AG, Ponomareva AA, Andrianova IA, Le Minh G, Rauova L, Litvinov RI
and Weisel JW: Platelet factor 4-containing immune complexes induce
platelet activation followed by calpain-dependent platelet death.
Cell Death Discov. 5:1062019. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Kulyar MF, Yao W, Ding Y, Du H, Li K,
Zhang L, Li A, Huachun P, Waqas M, Mehmood K and Li J: Cluster of
differentiation 147 (CD147) expression is linked with thiram
induced chondrocyte's apoptosis via Bcl-2/Bax/Caspase-3 signalling
in tibial growth plate under chlorogenic acid repercussion.
Ecotoxicol Environ Saf. 213:1120592021. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Ali N, Rashid S, Nafees S, Hasan SK,
Shahid A, Majed F and Sultana S: Protective effect of Chlorogenic
acid against methotrexate induced oxidative stress, inflammation
and apoptosis in rat liver: An experimental approach. Chem Biol
Interact. 272:80–91. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Yang C, Wang ZQ, Zhang ZC, Lou G and Jin
WL: CBL0137 activates ROS/BAX signaling to promote
caspase-3/GSDME-dependent pyroptosis in ovarian cancer cells.
Biomed Pharmacother. 161:1145292023. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhang Y, Yang X, Ge X and Zhang F:
Puerarin attenuates neurological deficits via Bcl-2/Bax/cleaved
caspase-3 and Sirt3/SOD2 apoptotic pathways in subarachnoid
hemorrhage mice. Biomed Pharmacother. 109:726–733. 2019. View Article : Google Scholar
|
|
77
|
Hu Q, Lyon CJ, Fletcher JK, Tang W, Wan M
and Hu TY: Extracellular vesicle activities regulating macrophage-
and tissue-mediated injury and repair responses. Acta Pharm Sin B.
11:1493–1512. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Wu MY, Liu L, Wang EJ, Xiao HT, Cai CZ,
Wang J, Su H, Wang Y, Tan J, Zhang Z, et al: PI3KC3 complex subunit
NRBF2 is required for apoptotic cell clearance to restrict
intestinal inflammation. Autophagy. 17:1096–1111. 2021. View Article : Google Scholar :
|
|
79
|
Kuo WT, Zuo L, Odenwald MA, Madha S, Singh
G, Gurniak CB, Abraham C and Turner JR: The tight junction protein
ZO-1 is dispensable for barrier function but critical for effective
mucosal repair. Gastroenterology. 161:1924–1939. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Kuo WT, Shen L, Zuo L, Shashikanth N, Ong
M, Wu L, Zha J, Edelblum KL, Wang Y, Wang Y, et al:
Inflammation-induced occludin downregulation limits epithelial
apoptosis by suppressing caspase-3 expression. Gastroenterology.
157:1323–1337. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Guo H, Guo H, Xie Y, Chen Y, Lu C, Yang Z,
Zhu Y, Ouyang Y, Zhang Y and Wang X: Mo(3)Se(4) nanoparticle with
ROS scavenging and multi-enzyme activity for the treatment of
DSS-induced colitis in mice. Redox Biol. 56:1024412022. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Wang R, Moniruzzaman M, Wong KY, Wiid P,
Harding A, Giri R, Tong WH, Creagh J, Begun J, McGuckin MA and
Hasnain SZ: Gut microbiota shape the inflammatory response in mice
with an epithelial defect. Gut Microbes. 13:1–18. 2021. View Article : Google Scholar
|
|
83
|
Bakheet SA, Alrwashied BS, Ansari MA,
Nadeem A, Attia SM, Assiri MA, Alqahtani F, Ibrahim KE and Ahmad
SF: CXCR3 antagonist AMG487 inhibits glucocorticoid-induced tumor
necrosis factor-receptor-related protein and inflammatory mediators
in CD45 expressing cells in collagen-induced arthritis mouse model.
Int Immunopharmacol. 84:1064942020. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Le HT, Golla K, Karimi R, Hughes MR,
Lakschevitz F, Cines DB, Kowalska MA, Poncz M, McNagny KM, Häkkinen
L and Kim H: Platelet factor 4 (CXCL4/PF4) upregulates matrix
metalloproteinase-2 (MMP-2) in gingival fibroblasts. Sci Rep.
12:186362022. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Kasirer-Friede A, Peuhu E, Ivaska J and
Shattil SJ: Platelet SHARPIN regulates platelet adhesion and
inflammatory responses through associations with αIIbβ3 and LUBAC.
Blood Adv. 6:2595–2607. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Yu G, Rux AH, Ma P, Bdeir K and Sachais
BS: Endothelial expression of E-selectin is induced by the
platelet-specific chemokine platelet factor 4 through LRP in an
NF-kappaB-dependent manner. Blood. 105:3545–3551. 2005. View Article : Google Scholar
|
|
87
|
Petrai I, Rombouts K, Lasagni L,
Annunziato F, Cosmi L, Romanelli RG, Sagrinati C, Mazzinghi B,
Pinzani M, Romagnani S, et al: Activation of p38(MAPK) mediates the
angiostatic effect of the chemokine receptor CXCR3-B. Int J Biochem
Cell Biol. 40:1764–1774. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Zeng B, Sun Z, Zhao Q, Liu D, Chen H, Li
X, Xing HR and Wang J: SEC23A inhibit melanoma metastatic through
secretory PF4 cooperation with SPARC to inhibit MAPK signaling
pathway. Int J Biol Sci. 17:3000–3012. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Bonacchi A, Romagnani P, Romanelli RG,
Efsen E, Annunziato F, Lasagni L, Francalanci M, Serio M, Laffi G,
Pinzani M, et al: Signal transduction by the chemokine receptor
CXCR3: Activation of Ras/ERK, Src, and phosphatidylinositol
3-kinase/Akt controls cell migration and proliferation in human
vascular pericytes. J Biol Chem. 276:9945–9954. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Van Raemdonck K, Gouwy M, Lepers SA, Van
Damme J and Struyf S: CXCL4L1 and CXCL4 signaling in human
lymphatic and microvascular endothelial cells and activated
lymphocytes: Involvement of mitogen-activated protein (MAP)
kinases, Src and p70S6 kinase. Angiogenesis. 17:631–640. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Li LX, Xia YT, Sun XY, Li LR, Yao L, Ali
MI, Gu W, Zhang JP, Liu J, Huang SG, et al: CXCL-10/CXCR3 in
macrophages regulates tissue repair by controlling the expression
of Arg1, VEGFa and TNFα. J Biol Regul Homeost Agents. 34:987–999.
2020.PubMed/NCBI
|