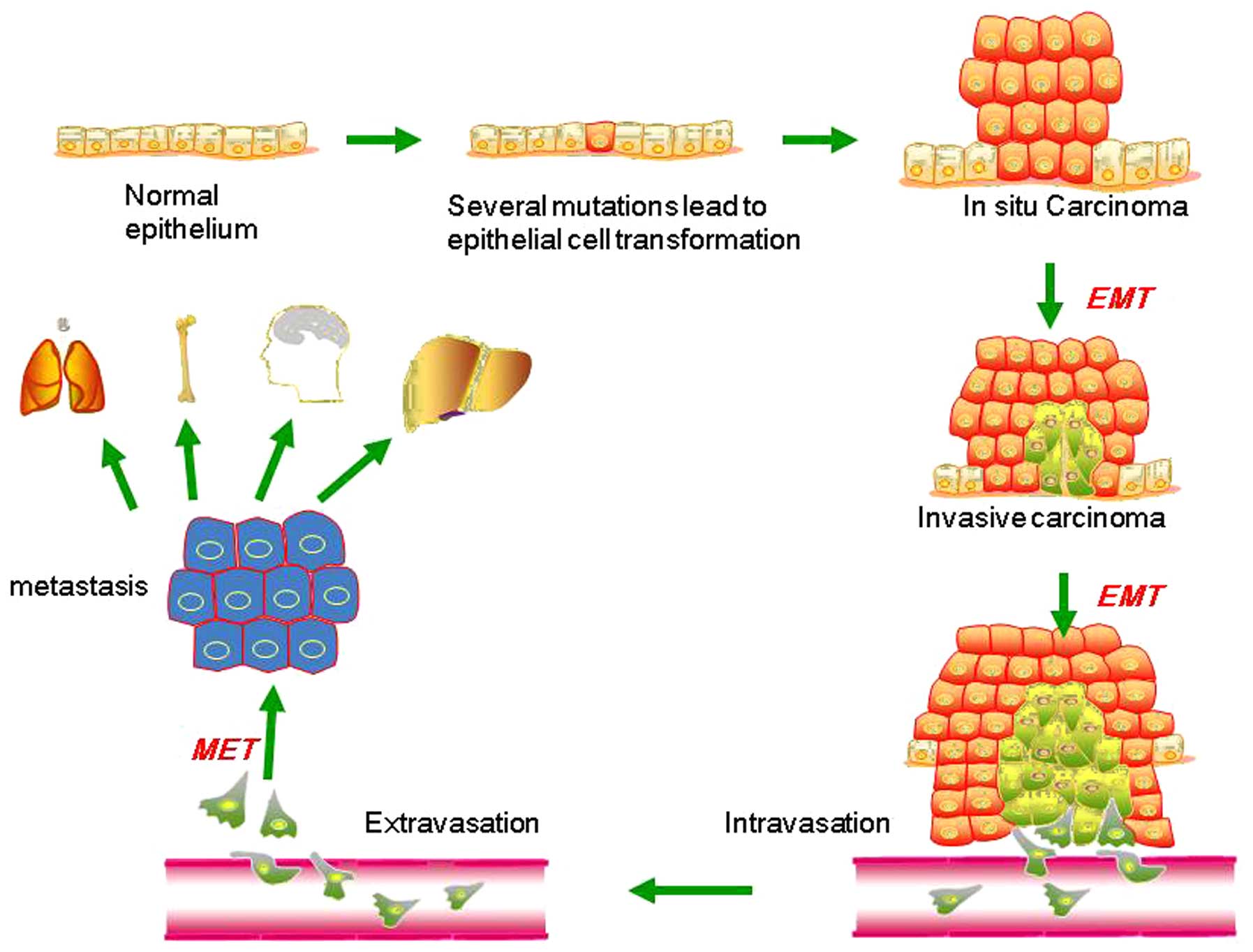
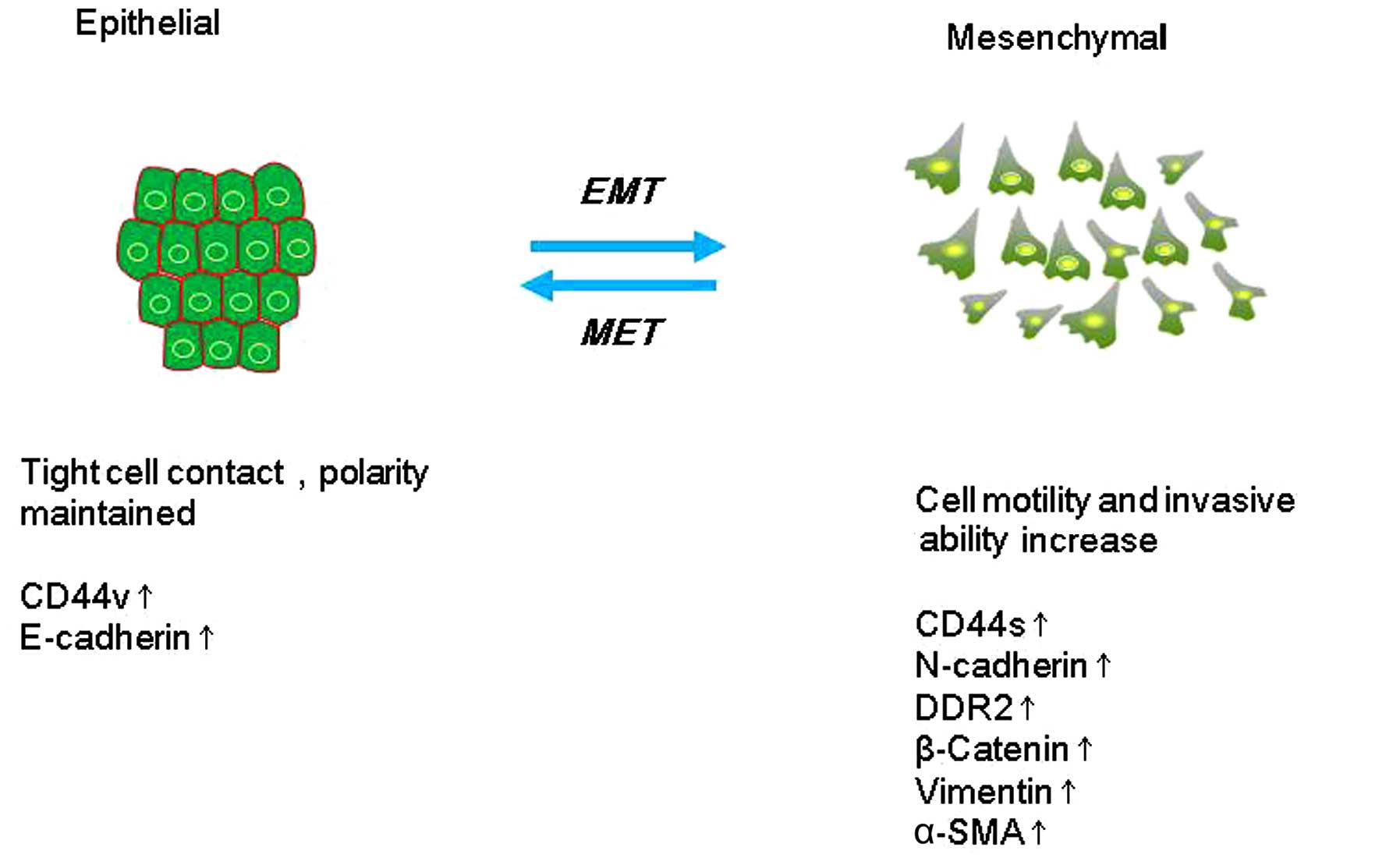
EMT describes a process in which cells lose
epithelial traits and gain mesenchymal characteristics. EMT is
characterized by loss of cell adhesion and phenotypic change from
typical cuboidal to an elongated spindle shape, leading to enhanced
migratory capacity (5). In the early
stage of tumor metastasis (Fig. 1),
cancer cells from the primary tumor could acquire invasive
properties and gain access to the blood or lymphatic vascular
systems as circulating tumor cells (CTCs) (6,7). This
procedure is aided by neo-angiogenesis and remodeling of the
basement membrane (8–10). In the bloodstream and lymphatic
vessels, CTCs are capable of surviving and eventually reach distant
secondary sites, including the bone, lungs, liver and brain. This
process is accomplished mainly by the mesenchymal-epithelial
transition (MET), which is a process opposite to the initial EMT at
the primary tumor site, and is considered to contribute
substantially to the colonization of CTCs into metastatic tumors at
the secondary site (11–14). Such dynamic EMT/MET state transitions
may play a critical role during tumor metastasis.
E-cadherin is a calcium-dependent cell-surface
protein that facilitates adhesion between epithelial cells
(15). E-cadherin is characterized by
long cytoplasmic and extracellular domains, which create homophilic
interactions between adjacent cells to facilitate adhesion
(16). A change in the expression of
E-cadherin is the typical epithelial cell marker of EMT (17). Suppression of E-cadherin function or
expression leads to mesenchymal morphology and increased cell
migration and invasion (18,19) as well as metastasis (20). In breast cancer, partial or total loss
of E-cadherin expression correlates with loss of differentiation
characteristics, acquisition of invasiveness, increased tumor
grade, metastatic behavior and poor prognosis (21–24).
Furthermore, its reduced expression can also be associated with
some non-lobular breast carcinomas of triple-negative phenotype
such as metaplastic carcinomas (25,26). Choi
et al analyses revealed that the loss of E-cadherin in
invasive carcinoma is greater than in pure ductal carcinoma in
situ (DCIS) in basal-like subtype cancer (27). Recently, changes in the level of
expression of different cadherins have been increasingly used to
monitor EMT. Indeed, the cadherin switch from E-cadherin to
N-cadherin has often been used to monitor the progress of EMT
during cancer progression. This switch increases cell motility and
the abilities of invasion and metastasis (28). In addition, E-cadherin also plays a
major role in the process of MET. In a previous study, Chao et
al reported the re-expression of E-cadherin at distant
metastatic tumors arising from E-cadherin-low or
E-cadherin-negative primary tumors (11). The authors reported strong E-cadherin
expression in >50% of liver, brain and lung metastases
originating from infiltrating ductal carcinoma of the breast
(29).
Cluster of differentiation (CD) 44 is a cell-surface
protein that modulates cellular signaling by forming co-receptor
complexes with various receptor tyrosine kinases. It plays an
important role in the metastasis of breast cancer (30). Some studies have shown an upregulation
of CD44 in a metastatic cell line as compared with a non-metastatic
cell line (31). There is a shift in
CD44 expression from the variant isoform (CD44v) to the standard
isoform (CD44s) during EMT. The splicing factor epithelial splicing
regulatory protein 1 could control the CD44 isoform switch, which
is critical for regulating the EMT phenotype. CD44s expression is
upregulated in high-grade human breast neoplasms, and is correlated
with the level of the mesenchymal marker N-cadherin in these tumors
(30).
Discoidin domain receptor 2 (DDR2) is an atypical
receptor tyrosine kinase. It is the collagen-specific receptor that
reflects adaptation to the altered ECM microenvironment associated
with the EMT (32). In adult tissues,
the expression of DDR2 is confined to subsets of fibroblasts or
vascular smooth muscle cells (33,34). In
cancer, DDR2 facilitates prostate cancer cells to adhere to type I
collagen. It plays an important role in prostate cancer bone
metastasis (35). In breast cancer,
DDR2 expression correlates with increased invasiveness, thus
demonstrating its utility in identifying EMT (36). It has been reported that activation of
the collagen I receptor DDR2 regulates Snail1 protein stability by
stimulating extracellular signal-regulated kinase 2 (ERK2)
activity. Activated ERK2 directly phosphorylates Snail1 and reduces
its ubiquitination; as a result, the half-life of Snail1 increases.
DDR2-mediated stabilization of Snail1 promotes breast cancer cell
invasion and metastasis in vivo (37). Recent research suggests that DDR2
facilitates breast cancer cell metastasis in vivo as well as
hypoxia-induced cell migration, invasion and EMT in vitro.
DDR2 could be a potential target to treat breast cancer metastasis
(38).
Vimentin is an intermediate filament that is used as
a marker of mesenchymal cells to distinguish them from epithelial
cells (41). Vimentin is expressed at
sites of cellular elongation, and is associated with a migratory
phenotype. Increased vimentin expression is frequently used as an
EMT marker in cancer (44,45). There is a positive correlation of
vimentin expression with augmented invasiveness and metastasis. In
breast cancer, it was observed that Smad-interacting protein 1
(SIP1) could regulate vimentin expression in epithelial breast
tumor cells, and that vimentin was distinctly related to SIP1
expression in invasive cell lines (46). These data suggest that the regulation
of vimentin by SIP1 can be independent of E-cadherin expression and
does not necessarily rely on the modulation of the β-catenin/TCF
pathway. Nevertheless, many other indirect mechanisms could be
involved (46). In addition, vimentin
expression was found to be significantly higher in patients with
stage IV disease, Bloom Richardson score 4 and progesterone
receptor negativity. That study revealed that vimentin expression
was a significant biomarker for predicting reduced disease-free
survival and overall survival in breast cancer (47).
The expression of E-cadherin plays a very important
role in the process of EMT. EMT transcription factors can be
classified based on their ability to repress E-cadherin directly or
indirectly.
The Snail family comprises three members, Snail 1, 2
and 3 (also termed Snail, Slug and Smuc, respectively). Snail 1 and
2 both repress the expression of the epithelial marker gene
cadherin 1 (CDH1), which encodes E-cadherin. Snail-induced EMT is
due to the direct repression of E-cadherin transcription. Snail is
the most widely recognized suppressor of E-cadherin expression
(52). Snail-induced EMT produces
concerted biophysical changes due to altered cytoskeletal gene
expression. Biophysical changes associated with cancer metastasis,
including elevated traction forces and loss of cytoskeletal and
nuclear structure, are directly induced by EMT in the absence of
any extraneous environmental cues (53). Snail promotes EMT in breast cancer
cells in part via activation of nuclear ERK2 (54). Furthermore, the overexpression of
Snail in MCF-7 breast cancer cells induced EMT, increased cell
migration, reduced cell adhesion and increased tumorigenicity
(54). In addition, the second member
of the Snail family, Slug, does play a major role during EMT. The
metastatic spread of triple-negative breast cancer may be
suppressed by blocking the Slug activity, which may specifically
inhibit the homing/colonization to the bone (55). However, the third member of the Snail
family, Smuc, does not play a major role during EMT (52).
The ZEB family (ZEB1/2) comprises zinc-finger
transcription factors that recognize a consensus E-box type
element, which are known as ZEB proteins (52). These proteins directly repress
E-cadherin expression independently of Snail transcription factors
in mouse mammary epithelial cells (56,57). ZEB1
and/or ZEB2 expression increased aggressiveness and metastatic
capacity in breast cancer (58). ZEB1
and ZEB2 not only repress E-cadherin but also other epithelial
markers involved in cell polarity, components of tight junctions,
gap junctions and desmosomes (59–61).
Moreover, ZEB1 plays an important role in tumor progression and
poor clinical outcomes in cancer patients. It is an specific EMT
inducer that dictates cancer stem cell properties, such as
radioresistance and drug resistance (62). ZEB2 directly represses the expression
of the tight junction proteins claudin-4 and zona occludens 3
(61). It also suppresses the
expression of the desmosome protein plakophilin-2, and induces the
expression of the mesenchymal proteins vimentin, N-cadherin and
matrix metalloproteinase-2 through a yet unknown mechanism
(61). It has been reported that
cytoplasmic ZEB2 is an important factor in the early stages of
malignancy, and it also predicts a poor overall survival rate in
invasive micropapillary carcinoma in a canine mammary cancer model
(58).
The members of the basic helix-loop-helix family
(Twist-1/2) are homodimers or heterodimers that can bind to a
consensus E-box sequence (63). Their
expression is upregulated during early embryonic morphogenesis,
tissue fibrosis and cancer metastasis (64–66).
Overexpression of Twist produced a transformation of the MCF-7 cell
line that exhibited many of the traits representative of an
epithelial-mesenchymal-like transition (67). In addition, we also reported that
Twist was able to upregulate vascular endothelial growth factor
(VEGF) synthesis and to induce in vivo angiogenesis
(67). Expression of Twist-1 is
associated with poor survival in carcinoma. However, the potential
of Twist-1 as a therapeutic target in cancer treatment still
requires validation in further research (68).
The other group of CDH1 repressors (indirect
regulators) comprises Forkhead box protein C2 (FoxC2), goosecoid,
TCF4 and X-box binding protein 1 (XBP1) (69–72). FoxC2
is a winged helix/forkhead domain transcription factor that acts as
a pleiotropic inducer of EMT. FoxC2 is expressed in ductal breast
cancers and metastatic breast cancer cell lines (69). Elevated levels of FoxC2 protein were
associated with the basal-like breast cancer phenotype and with a
poor rate of disease-free survival (73). In addition, expression of FoxC2 and
co-expression of Twist and FoxC2 in the stroma of breast phyllodes
tumors contributed to poorer prognosis (74). The goosecoid homeobox transcription
factor is overexpressed in the majority of human breast tumors.
Ectopic expression of goosecoid in human breast cells caused
invasion-associated cellular changes, including EMT. Moreover,
goosecoid significantly enhanced the ability of breast cancer cells
to form pulmonary metastases in mice (75). TCF4 belongs to the β-catenin pathway,
and is one of the ZEB family transcription factors. TCF-4-regulated
osteopontin (OPN) expression and cell invasion may be dependent on
Wnt signaling activity, and TCF-4 and OPN may become a novel
prognostic indicator in breast cancer when considered together
(71). Moreover, XBP1 is an important
transcription factor within the cAMP response element binding
protein/activating transcription factor family, and it contains a
basic leucine zipper structure (72).
It has been reported that the forced expression of XBP1 induces EMT
in breast cancer cells. The authors specified that XBP1 decreases
the expression of the epithelial marker E-cadherin but increases
the expression of the mesenchymal markers N-cadherin and vimentin.
These results indicated that XBP1 induces EMT and cell invasion in
breast cancer cells by promoting Snail expression (72).
miRNAs, functioning as co-activators or
co-repressors, are key players in cell plasticity, being
specifically involved in cell regulation with EMT-related
transcription factors. miRNAs that contribute to EMT in breast
cancer are categorized as either EMT inducers or EMT repressors
(Table I).
The well-known oncomiR miR-21 was identified as an
EMT inducer, while phosphatase and tensin homolog (PTEN) is a major
miR-21 target that negatively regulates EMT phenotypes (76). The antagonism of miR-21 in the
MDA-MB-231 aggressive breast cancer cell line is found to reverse
EMT, which is accompanied by PTEN upregulation and AKT/ERK1/2
inactivation (77). That study
suggests that miR-21 functions as an oncogene and modulates
tumorigenesis, and it may serve as a novel therapeutic target.
miR-10b was identified as a positive regulator of
EMT, and it was demonstrated to be a positive effector of Twist. It
was shown to induce migration and invasion capacities in breast
cancer cells via direct targeting of homeobox D10 (HOXD10)
transcription (78). miR-10b-mediated
suppression of HOXD10 has also been shown to promote the expression
of Ras homolog family member C, which leads to cell invasion and
migration in the non-metastatic breast cancer cell line SUM149.
Moreover, downregulation of miR-10b reduces the metastatic burden
of breast cancer in vivo (79).
miR-9 is upregulated in breast cancer cells, and
directly targets CDH1, leading to increased cell motility and
invasiveness (80). Downregulation of
miR-9-mediated E-cadherin expression results in the activation of
β-catenin signaling, which contributes to the upregulation of VEGF;
in turn, this leads to increased tumor angiogenesis (80). Overexpression of miR-9 is also found
in tumors with aggressive phenotypes, and is related to poor
prognosis in breast cancer, suggesting that it may serve as a
potential biomarker for the progression of breast cancer as well as
a target for treatment (81).
miR-103/107 inhibit the expression of Dicer, causing
global miRNA downregulation. In breast cancer, high levels of
miR-103/107 are associated with metastasis and poor outcome.
miR-103/107 confer migratory capacities in vitro and empower
metastatic dissemination of otherwise non-aggressive cells in
vivo (82). Inhibition of
miR-103/107 prevents migration and metastasis of malignant
cells.
The miR-200 family members (miR-200a, miR- 200b,
miR-200c, miR-141 and miR-429) were identified as the guardians of
the epithelial phenotype in breast cancer (83). The miR-200 family activates the
Sec23a-mediated tumor cell secretome, which leads to the secretion
of metastasis-suppressive proteins (84). Predictably, loss of miRNA-200a is
frequently observed in breast cancer, but this loss does not
predict tumor recurrence or patient survival (85). miR-200 family members are encoded from
two clusters, and directly target the messenger RNAs of the
E-cadherin transcriptional repressors ZEB1 and ZEB2 (84). Notably, Burk et al and other
studies (83,86) have shown that both promoter regions
are repressed in mesenchymal cells by ZEB1 and ZEB2 through binding
to a conserved pair of ZEB-type E-box elements. These studies
established the existence of a double-negative feedback loop
controlling ZEB1-ZEB2 and miR-200 family expression.
miR-375 is elevated in epithelial-like breast cancer
cells, and ectopic miR-375 expression suppresses EMT in
mesenchymal-like breast cancer cells. The authors identified short
stature homeobox 2 (SHOX2) as a miR-375 target, and
miR-375-mediated suppression in EMT was reversed by forced SHOX2
expression. This study reveals that the association between miR-375
and SHOX2 is a potent EMT regulator and plays a critical role in
breast tumorigenicity (87).
miR-506, which is a novel miRNA, was found to be
significantly related to breast cancer patient survival. It
suppressed the expression of mesenchymal markers in the MDA-MB-231
human breast cancer cell line. In addition, nuclear factor-κB
(NF-κB) combines with the upstream promoter region of miR-506 to
suppress transcription (87). It
inhibited transforming growth factor (TGF)-β-induced EMT and
suppressed adhesion, invasion and migration of MDA-MB-231 cells
when miR-506 was overexpressed (88).
In general, miR-506 plays a key role in the process of EMT via
post-translational control of EMT-related genes.
During Snail-induced EMT in MCF7 breast cancer
cells, miR-203 is repressed in a correlated manner. In particular,
miR-203 represses endogenous Snail, forming a double-negative
miR-203/Snail feedback loop (89). In
addition, miR-203 is also able to target Slug. TGF-β induces Slug
to promote EMT by repressing the miR-203 promoter to inhibit its
transcription. It was found that miR-203 is significantly
downregulated in highly metastatic breast cancer cells, and that
the restoration of miR-203 in these cells inhibits tumor cell
invasion in vitro and lung metastatic colonization in
vivo by repressing Slug (90).
miR-34 is one of the most studied tumor-suppressor
miRNAs. It is implicated in the inhibition of EMT mediated by p53.
It was reported that activation of p53 downregulates the EMT
induced by the transcription factor Snail via induction of the
miR-34 gene. Suppression of miR-34 caused upregulation of Snail and
EMT markers, and enhanced cell migration and invasion. Moreover,
miR-34a prevents TGF-β-induced EMT, and the repression of the
miR-34 gene by Snail and related factors is part of the EMT program
(91).
Several kinases regulate EMT, stemness or
metastasis, including FYN proto-oncogene, Src family tyrosine
kinase, platelet-derived growth factor receptor α, BRAF,
Fms-related tyrosine kinase 1, LYN proto-oncogene, Src family
tyrosine kinase and YES proto-oncogene 1, Src family tyrosine
kinase (92–94). In the present report, we review two
recent studies on breast cancer occurrence and EMT-associated
protein kinases.
AXL receptor tyrosine kinase (AXL) is a member of
the Tyro3-AXL-Mer family of receptor tyrosine kinases. AXL is
overexpressed in a wide variety of human cancers, with significant
correlation with tumor stage in breast cancer patients. It plays a
important role in cancer progression and metastasis (95–97).
Asiedu et al (98) reported
that AXL overexpression in HMLE cells downregulated E-cadherin,
while the expression of mesenchymal markers such as N-cadherin,
Snail and Slug was upregulated. A similar result was observed by
forced expression of AXL in the normal human mammary epithelial
cell line MCF10A. On the contrary, the silencing of AXL by
viral-mediated shRNA led to the upregulation of epithelial markers,
while mesenchymal markers were downregulated. Inactivation of AXL
also led to downregulation of the NF-κB pathway and reduced tumor
formation in vivo (98). In
addition, AXL is overexpressed in highly invasive breast cancer
cell lines. By contrast, weakly invasive breast cancer cell lines
do not or merely express limited quantities of AXL. AXL may be an
important therapeutic target in inflammatory breast cancer
(99).
CDKL2 is one of the most distant members of the cell
division cycle protein 2-related serine/threonine protein kinase
and mitogen-activated protein kinase family, which is also known as
p56 or KKIAMRE (100,101). A recent study (100) demonstrated that CDKL2 activated a
positive feedback loop, consisting of ZEB1/E-cadherin/β-catenin, to
induce EMT in breast cancer. As a result, E-cadherin expression was
reduced, and the epithelial barrier was broken down, which led to
nuclear translocation of β-catenin as well as elevated
β-catenin/TCF4 transcriptional activity. Activated β-catenin
increased ZEB1 promoter activity and transcription, which in turn
resulted in further suppression of E-cadherin expression and
continuous activation of the positive feedback loop (100) This result suggested that CDKL2 can
be a potential prognostic factor for poor outcome and a therapeutic
target for human invasive breast cancer.
Currently, the role of EMT and MET in breast tumors
is under investigation, and the molecular mechanism of EMT and MET
is being revealed. With the development of individualized breast
cancer therapies, new prognostic and predictive biomarkers are
required to facilitate clinical decision-making processes. Further
studies on the link between EMT markers and breast cancer, such as
the link between EMT and biomarkers, the regulatory association
between transcription factors and miRNAs, and the link between EMT
and protein kinases, will contribute to the identification of
biomarkers for predicting early breast cancer metastasis and to
identify intervention therapeutic targets in breast cancer, as well
as to provide new ideas and methods for the treatment of breast
cancer.
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
DeSantis C, Siegel R, Bandi P and Jemal A:
Breast cancer statistics. CA Cancer J Cli. 61:409–418. 2011.
|
|
3
|
O'Shaughnessy J: Extending survival with
chemotherapy in metastatic breast cancer. Oncologist. 10:(Suppl 3).
S20–S29. 2005. View Article : Google Scholar
|
|
4
|
Creighton CJ, Chang JC and Rosen JM:
Epithelial-mesenchymal transition (EMT) in tumor-initiating cells
and its clinical implications in breast cancer. J Mammary Gland
Biol Neoplasia. 15:253–260. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Guarino M, Rubino B and Ballabio G: The
role of epithelial-mesenchymal transition in cancer pathology.
Pathology. 39:305–318. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Woodhouse EC, Chuaqui RF and Liotta LA:
General mechanisms of metastasis. Cancer 80 (Suppl). 1529–1537.
1997. View Article : Google Scholar
|
|
7
|
Chambers AF, Groom AC and MacDonald IC:
Dissemination and growth of cancer cells in metastatic sites. Nat
Rev Cancer. 2:563–572. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Weidner N, Folkman J, Pozza F, Bevilacqua
P, Allred EN, Moore DH, Meli S and Gasparini G: Tumor angiogenesis:
A new significant and independent prognostic indicator in
early-stage breast carcinoma. J Natl Cancer Ins. 84:1875–1887.
1992. View Article : Google Scholar
|
|
9
|
Folkman J and Shing Y: Angiogenesis. J
Biol chem. 267:10931–10934. 1992.PubMed/NCBI
|
|
10
|
Folkman J: The role of angiogenesis in
tumor growth. S Cancer Biol. 3:65–71. 1992.
|
|
11
|
Chao YL, Shepard CR and Wells A: Breast
carcinoma cells re-express E-cadherin during mesenchymal to
epithelial reverting transition. Mol Cancer. 9:1792010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chaffer CL, Brennan JP, Slavin JL, Blick
T, Thompson EW and Williams ED: Mesenchymal-to-epithelial
transition facilitates bladder cancer metastasis: Role of
fibroblast growth factor receptor-2. Cancer Res. 66:11271–11278.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chaffer CL, Thompson EW and Williams ED:
Mesenchymal to epithelial transition in development and disease.
Cells Tissues Organs. 185:7–19. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hugo H, Ackland ML, Blick T, Lawrence MG,
Clements JA, Williams ED and Thompson EW: Epithelial-mesenchymal
and mesenchymal-epithelial transitions in carcinoma progression. J
Cell Physiol. 213:374–383. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hyafil F, Babinet C and Jacob F: Cell-cell
interactions in early embryogenesis: A molecular approach to the
role of calcium. Cell. 26:447–454. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cavallaro U and Christofori G: Cell
adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev
Cancer. 4:118–132. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hay ED and Zuk A: Transformations between
epithelium and mesenchyme: Normal, pathological, and experimentally
induced. Am J Kidney Dis. 26:678–690. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vleminckx K, Vakaet L Jr, Mareel M, Fiers
W and van Roy F: Genetic manipulation of E-cadherin expression by
epithelial tumor cells reveals an invasion suppressor role. Cell.
66:107–119. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Perl AK, Wilgenbus P, Dahl U, Semb H and
Christofori G: A causal role for E-cadherin in the transition from
adenoma to carcinoma. Nature. 392:190–193. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Canel M, Serrels A, Frame MC and Brunton
VG: E-cadherin-integrin crosstalk in cancer invasion and
metastasis. J Cell Sci. 126:393–401. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Heimann R, Lan F, McBride R and Hellman S:
Separating favorable from unfavorable prognostic markers in breast
cancer: The role of E-cadherin. Cancer Res. 60:298–304.
2000.PubMed/NCBI
|
|
22
|
Hunt NC, Douglas-Jones AG, Jasani B,
Morgan JM and Pignatelli M: Loss of E-cadherin expression
associated with lymph node metastases in small breast carcinomas.
Virchows Arch. 430:285–289. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Oka H, Shiozaki H, Kobayashi K, Inoue M,
Tahara H, Kobayashi T, Takatsuka Y, Matsuyoshi N, Hirano S and
Takeichi M: Expression of E-cadherin cell adhesion molecules in
human breast cancer tissues and its relationship to metastasis.
Cancer Res. 53:1696–1701. 1993.PubMed/NCBI
|
|
24
|
Siitonen SM, Kononen JT, Helin HJ, Rantala
IS, Holli KA and Isola JJ: Reduced E-cadherin expression is
associated with invasiveness and unfavorable prognosis in breast
cancer. Am J Clin Pathol. 105:394–402. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mahler-Araujo B, Savage K, Parry S and
Reis-Filho JS: Reduction of E-cadherin expression is associated
with non-lobular breast carcinomas of basal-like and triple
negative phenotype. J Clin Pathol. 61:615–620. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lopes N, Carvalho J, Durães C, Sousa B,
Gomes M, Costa JL, Oliveira C, Paredes J and Schmitt F: 1Alpha,
25-dihydroxyvitamin D3 induces de novo E-cadherin expression in
triple-negative breast cancer cells by CDH1-promoter demethylation.
Anticancer Res. 32:249–257. 2012.PubMed/NCBI
|
|
27
|
Choi Y, Lee HJ, Jang MH, Gwak JM, Lee KS,
Kim EJ, Kim HJ, Lee HE and Park SY: Epithelial-mesenchymal
transition increases during the progression of in situ to invasive
basal-like breast cancer. Hum Pathol. 44:2581–2589. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gravdal K, Halvorsen OJ, Haukaas SA and
Akslen LA: A switch from E-cadherin to N-cadherin expression
indicates epithelial to mesenchymal transition and is of strong and
independent importance for the progress of prostate cancer. Clin
Cancer Res. 13:7003–7011. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chao Y, Wu Q, Acquafondata M, Dhir R and
Wells A: Partial mesenchymal to epithelial reverting transition in
breast and prostate cancer metastases. Cancer Microenviron:.
5:19–28. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Brown RL, Reinke LM, Damerow MS, Perez D,
Chodosh LA, Yang J and Cheng C: CD44 splice isoform switching in
human and mouse epithelium is essential for epithelial-mesenchymal
transition and breast cancer progression. J Clin Invest.
121:1064–1074. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Leth-Larsen R, Lund R, Hansen HV,
Laenkholm AV, Tarin D, Jensen ON and Ditzel HJ: Metastasis-related
plasma membrane proteins of human breast cancer cells identified by
comparative quantitative mass spectrometry. Mol Cell Proteomics.
8:1436–1449. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vogel W, Gish GD, Alves F and Pawson T:
The discoidin domain receptor tyrosine kinases are activated by
collagen. Mol Cell. 1:13–23. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zeisberg EM, Tarnavski O, Zeisberg M,
Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT,
Roberts AB, et al: Endothelial-to-mesenchymal transition
contributes to cardiac fibrosis. Nat Med. 13:952–961. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Goldsmith EC, Hoffman A, Morales MO, Potts
JD, Price RL, McFadden A, Rice M and Borg TK: Organization of
fibroblasts in the heart. Dev Dyn. 230:787–794. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yan Z, Jin S, Wei Z, Huilian H, Zhanhai Y,
Yue T, Juan L, Jing L, Libo Y and Xu L: Discoidin domain receptor 2
facilitates prostate cancer bone metastasis via regulating
parathyroid hormone-related protein. Biochim Biophys Acta.
1842:1350–1363. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Evtimova V, Zeillinger R and Weidle UH:
Identification of genes associated with the invasive status of
human mammary carcinoma cell lines by transcriptional profiling.
Tumor Biol. 24:189–198. 2003. View Article : Google Scholar
|
|
37
|
Zhang K, Corsa CA, Ponik SM, Prior JL,
Piwnica-Worms D, Eliceiri KW, Keely PJ and Longmore GD: The
collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to
facilitate breast cancer metastasis. Nat Cell Biol. 15:677–687.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ren T, Zhang W, Liu X, Zhao H, Zhang J,
Zhang J, Li X, Zhang Y, Bu X, Shi M, et al: Discoidin domain
receptor 2 (DDR2) promotes breast cancer cell metastasis and the
mechanism implicates epithelial-mesenchymal transition programme
under hypoxia. J Pathol. 234:526–537. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bienz M: beta-Catenin: A pivot between
cell adhesion and Wnt signalling. Curr Biol. 15:R64–R67. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Brabletz T, Jung A, Hermann K, Günther K,
Hohenberger W and Kirchner T: Nuclear overexpression of the
oncoprotein beta-catenin in colorectal cancer is localized
predominantly at the invasion front. Pathol Res Pract. 194:701–704.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zeisberg M and Neilson EG: Biomarkers for
epithelial-mesenchymal transitions. J Clin Invest. 119:1429–1437.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Prasad CP, Rath G, Mathur S, Bhatnagar D,
Parshad R and Ralhan R: Expression analysis of E-cadherin, Slug and
GSK3beta in invasive ductal carcinoma of breast. BMC Cancer.
9:3252009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li L, Liu C, Amato RJ, Chang JT, Du G and
Li W: CDKL2 promotes epithelial-mesenchymal transition and breast
cancer progression. Oncotarget. 5:10840–10853. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Scanlon CS, Van Tubergen EA, Inglehart RC
and D'Silva NJ: Biomarkers of epithelial-mesenchymal transition in
squamous cell carcinoma. J Dent Res. 92:114–121. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Raymond WA and Leong AS: Vimentin - a new
prognostic parameter in breast carcinoma? J Pathol. 158:107–114.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bindels S, Mestdagt M, Vandewalle C,
Jacobs N, Volders L, Noël A, van Roy F, Berx G, Foidart JM and
Gilles C: Regulation of vimentin by SIP1 in human epithelial breast
tumor cells. Oncogene. 25:4975–4985. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Patel NA, Patel PS and Vora HH: Role of
PRL-3, Snail, Cytokeratin and Vimentin expression in epithelial
mesenchymal transition in breast carcinoma. Breast Dis. 35:113–127.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gabbiani G, Kapanci Y, Barazzone P and
Franke WW: Immunochemical identification of intermediate-sized
filaments in human neoplastic cells. A diagnostic aid for the
surgical pathologist. Am J Pathol. 104:206–216. 1981.PubMed/NCBI
|
|
49
|
Damonte P, Gregg JP, Borowsky AD, Keister
BA and Cardiff RD: EMT tumorigenesis in the mouse mammary gland.
Lab Invest. 87:1218–1226. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sarrió D, Rodriguez-Pinilla SM, Hardisson
D, Cano A, Moreno-Bueno G and Palacios J: Epithelial-mesenchymal
transition in breast cancer relates to the basal-like phenotype.
Cancer Res. 68:989–997. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yamashita M, Ogawa T, Zhang X, Hanamura N,
Kashikura Y, Takamura M, Yoneda M and Shiraishi T: Role of stromal
myofibroblasts in invasive breast cancer: Stromal expression of
alpha-smooth muscle actin correlates with worse clinical outcome.
Breast Cancer. 19:170–176. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Barrallo-Gimeno A and Nieto MA: The Snail
genes as inducers of cell movement and survival: Implications in
development and cancer. Development. 132:3151–3161. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
McGrail DJ, Mezencev R, Kieu QM, McDonald
JF and Dawson MR: SNAIL-induced epithelial-to-mesenchymal
transition produces concerted biophysical changes from altered
cytoskeletal gene expression. FASEB. 29:1280–1289. 2015. View Article : Google Scholar
|
|
54
|
Smith BN, Burton LJ, Henderson V, Randle
DD, Morton DJ, Smith BA, Taliaferro-Smith L, Nagappan P, Yates C,
Zayzafoon M, et al: Snail promotes epithelial mesenchymal
transition in breast cancer cells in part via activation of nuclear
ERK2. PloS One. 9:e1049872014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ferrari-Amorotti G, Chiodoni C, Shen F,
Cattelani S, Soliera AR, Manzotti G, Grisendi G, Dominici M, Rivasi
F, Colombo MP, et al: Suppression of invasion and metastasis of
triple-negative breast cancer lines by pharmacological or genetic
inhibition of slug activity. Neoplasia. 16:1047–1058. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Eger A, Aigner K, Sonderegger S, Dampier
B, Oehler S, Schreiber M, Berx G, Cano A, Beug H and Foisner R:
DeltaEF1 is a transcriptional repressor of E-cadherin and regulates
epithelial plasticity in breast cancer cells. Oncogene.
24:2375–2385. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Shirakihara T, Saitoh M and Miyazono K:
Differential regulation of epithelial and mesenchymal markers by
deltaEF1 proteins in epithelial mesenchymal transition induced by
TGF-beta. Mol Biol Cell. 18:3533–3544. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Gamba CO, Campos LC, Negreiros-Lima GL,
Maciel-Lima K, Sousa LP, Estrela-Lima A, Ferreira E and Cassali GD:
ZEB2 and ZEB1 expression in a spontaneous canine model of invasive
micropapillary carcinoma of the mammary gland. Res Vet Sci.
97:554–559. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Aigner K, Dampier B, Descovich L, Mikula
M, Sultan A, Schreiber M, Mikulits W, Brabletz T, Strand D, Obrist
P, et al: The transcription factor ZEB1 (deltaEF1) promotes tumor
cell dedifferentiation by repressing master regulators of
epithelial polarity. Oncogene. 26:6979–6988. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Aigner K, Descovich L, Mikula M, Sultan A,
Dampier B, Bonné S, van Roy F, Mikulits W, Schreiber M, Brabletz T,
et al: The transcription factor ZEB1 (deltaEF1) represses
Plakophilin 3 during human cancer progression. FEBS Lett.
581:1617–1624. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Vandewalle C, Comijn J, De Craene B,
Vermassen P, Bruyneel E, Andersen H, Tulchinsky E, Van Roy F and
Berx G: SIP1/ZEB2 induces EMT by repressing genes of different
epithelial cell-cell junctions. Nucleic Acids Res. 33:6566–6578.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhang P, Sun Y and Ma L: ZEB1: At the
crossroads of epithelial-mesenchymal transition, metastasis and
therapy resistance. Cell Cycle. 14:481–487. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Moyret-Lalle C, Ruiz E and Puisieux A:
Epithelial-mesenchymal transition transcription factors and miRNAs:
‘Plastic surgeons’ of breast cancer. World J Clin Oncol. 5:311–322.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Yu W, Kamara H and Svoboda KK: The role of
twist during palate development. Dev Dyn. 237:2716–2725. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kida Y, Asahina K, Teraoka H, Gitelman I
and Sato T: Twist relates to tubular epithelial-mesenchymal
transition and interstitial fibrogenesis in the obstructed kidney.
J Histochem Cytochem. 55:661–673. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Yang MH, Wu MZ, Chiou SH, Chen PM, Chang
SY, Liu CJ, Teng SC and Wu KJ: Direct regulation of TWIST by
HIF-1alpha promotes metastasis. Nat Cell Biol. 10:295–305. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Mironchik Y, Winnard PT Jr, Vesuna F, Kato
Y, Wildes F, Pathak AP, Kominsky S, Artemov D, Bhujwalla Z, Van
Diest P, et al: Twist overexpression induces in vivo angiogenesis
and correlates with chromosomal instability in breast cancer.
Cancer Res. 65:10801–10809. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Wushou A, Hou J, Zhao YJ and Shao ZM:
Twist-1 up-regulation in carcinoma correlates to poor survival. Int
J Mol Sci. 15:21621–21630. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Mani SA, Yang J, Brooks M, Schwaninger G,
Zhou A, Miura N, Kutok JL, Hartwell K, Richardson AL and Weinberg
RA: Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis
and is associated with aggressive basal-like breast cancers. Proc
Natl Acad Sci USA. 104:10069–10074. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Taube JH, Herschkowitz JI, Komurov K, et
al: Core epithelial-to-mesenchymal transition interactome
gene-expression signature is associated with claudin-low and
metaplastic breast cancer subtypes. Proceedings of the National
Academy of Sciences of the United States of America.
107:15449–15454. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Ravindranath A, Yuen HF, Chan KK, Grills
C, Fennell DA, Lappin TR and El-Tanani M: Wnt-β-catenin-Tcf-4
signalling-modulated invasiveness is dependent on osteopontin
expression in breast cancer. Br J Cancer. 105:542–551. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Li H, Chen X, Gao Y, Wu J, Zeng F and Song
F: XBP1 induces snail expression to promote epithelial-
to-mesenchymal transition and invasion of breast cancer cells. Cell
Signal. 27:82–89. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Li Y, Yang W, Yang Q and Zhou S: Nuclear
localization of GLI1 and elevated expression of FOXC2 in breast
cancer is associated with the basal-like phenotype. Histol
Histopathol. 27:475–484. 2012.PubMed/NCBI
|
|
74
|
Lim JC, Koh VC, Tan JS, Tan WJ, Thike AA
and Tan PH: Prognostic significance of epithelial-mesenchymal
transition proteins Twist and Foxc2 in phyllodes tumors of the
breast. Breast Cancer Res Treat. 150:19–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Hartwell KA, Muir B, Reinhardt F,
Carpenter AE, Sgroi DC and Weinberg RA: The Spemann organizer gene,
Goosecoid, promotes tumor metastasis. Proc Natl Acad Sci USA.
103:18969–18974. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Si ML, Zhu S, Wu H, Lu Z, Wu F and Mo YY:
miR-21-mediated tumor growth. Oncogene. 26:2799–2803. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Han M, Liu M, Wang Y, Chen X, Xu J, Sun Y,
Zhao L, Qu H, Fan Y and Wu C: Antagonism of miR-21 reverses
epithelial-mesenchymal transition and cancer stem cell phenotype
through AKT/ERK1/2 inactivation by targeting PTEN. PloS One.
7:e395202012. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Harquail J, Benzina S and Robichaud GA:
MicroRNAs and breast cancer malignancy: An overview of
miRNA-regulated cancer processes leading to metastasis. Cancer
Biomark. 11:269–280. 2012.PubMed/NCBI
|
|
79
|
Ma L, Reinhardt F, Pan E, Soutschek J,
Bhat B, Marcusson EG, Teruya-Feldstein J, Bell GW and Weinberg RA:
Therapeutic silencing of miR-10b inhibits metastasis in a mouse
mammary tumor model. Nat Biotechnol. 28:341–347. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Ma L, Young J, Prabhala H, Pan E, Mestdagh
P, Muth D, Teruya-Feldstein J, Reinhardt F, Onder TT, Valastyan S,
et al: miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin
and cancer metastasis. Nat Cell Biol. 12:247–256. 2010.PubMed/NCBI
|
|
81
|
Gwak JM, Kim HJ, Kim EJ, Chung YR, Yun S,
Seo AN, Lee HJ and Park SY: MicroRNA-9 is associated with
epithelial-mesenchymal transition, breast cancer stem cell
phenotype and tumor progression in breast cancer. Breast Cancer Res
Treat. 147:39–49. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Martello G, Rosato A, Ferrari F, Manfrin
A, Cordenonsi M, Dupont S, Enzo E, Guzzardo V, Rondina M, Spruce T,
et al: A MicroRNA targeting dicer for metastasis control. Cell.
141:1195–1207. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Burk U, Schubert J, Wellner U, Schmalhofer
O, Vincan E, Spaderna S and Brabletz T: A reciprocal repression
between ZEB1 and members of the miR-200 family promotes EMT and
invasion in cancer cells. EMBO Rep. 9:582–589. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Korpal M, Ell BJ, Buffa FM, Ibrahim T,
Blanco MA, Celià-Terrassa T, Mercatali L, Khan Z, Goodarzi H, Hua
Y, et al: Direct targeting of Sec23a by miR-200s influences cancer
cell secretome and promotes metastatic colonization. Nat Med.
17:1101–1108. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Jang K, Ahn H, Sim J, Han H, Abdul R, Paik
SS, Chung MS and Jang SJ: Loss of microRNA-200a expression
correlates with tumor progression in breast cancer. Transl Res.
163:242–251. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Bracken CP, Gregory PA, Kolesnikoff N,
Bert AG, Wang J, Shannon MF and Goodall GJ: A double-negative
feedback loop between ZEB1-SIP1 and the microRNA-200 family
regulates epithelial-mesenchymal transition. Cancer Res.
68:7846–7854. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Hong S, Noh H, Teng Y, Shao J, Rehmani H,
Ding HF, Dong Z, Su SB, Shi H, Kim J and Huang S: SHOX2 is a direct
miR-375 target and a novel epithelial-to-mesenchymal transition
inducer in breast cancer cells. Neoplasia. 16:279–290.e1-5. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Arora H, Qureshi R and Park WY: Mir-506
regulates epithelial mesenchymal transition in breast cancer cell
lines. PloS One. 8:e642732013. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Moes M, Le Bechec A, Crespo I, Laurini C,
Halavatyi A, Vetter G, Del Sol A and Friederich E: A novel network
integrating a miRNA-203/SNAI1 feedback loop which regulates
epithelial to mesenchymal transition. PloS One. 7:e354402012.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Ding X, Park SI, McCauley LK and Wang CY:
Signaling between transforming growth factor β(TGF-β) and
transcription factor SNAI2 represses expression of microRNA miR-203
to promote epithelial-mesenchymal transition and tumor metastasis.
J Biol Chem. 288:10241–10253. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Siemens H, Jackstadt R, Hünten S, Kaller
M, Menssen A, Götz U and Hermeking H: miR-34 and SNAIL form a
double-negative feedback loop to regulate epithelial-mesenchymal
transitions. Cell Cycle. 10:4256–4271. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Saito YD, Jensen AR, Salgia R and Posadas
EM: Fyn: A novel molecular target in cancer. Cancer. 116:1629–1637.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Jechlinger M, Sommer A, Moriggl R, Seither
P, Kraut N, Capodiecci P, Donovan M, Cordon-Cardo C, Beug H and
Grünert S: Autocrine PDGFR signaling promotes mammary cancer
metastasis. J Clin Invest. 116:1561–1570. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Choi YL, Bocanegra M, Kwon MJ, Shin YK,
Nam SJ, Yang JH, Kao J, Godwin AK and Pollack JR: LYN is a mediator
of epithelial-mesenchymal transition and a target of dasatinib in
breast cancer. Cancer Res. 70:2296–2306. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Hutterer M, Knyazev P, Abate A, Reschke M,
Maier H, Stefanova N, Knyazeva T, Barbieri V, Reindl M, Muigg A, et
al: Axl and growth arrest-specific gene 6 are frequently
overexpressed in human gliomas and predict poor prognosis in
patients with glioblastoma multiforme. Clin Cancer Res. 14:130–138.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Li Y, Ye X, Tan C, Hongo JA, Zha J, Liu J,
Kallop D, Ludlam MJ and Pei L: Axl as a potential therapeutic
target in cancer: Role of Axl in tumor growth, metastasis and
angiogenesis. Oncogene. 28:3442–3455. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Zhang YX, Knyazev PG, Cheburkin YV, Sharma
K, Knyazev YP, Orfi L, Szabadkai I, Daub H, Kéri G and Ullrich A:
AXL is a potential target for therapeutic intervention in breast
cancer progression. Cancer Res. 68:1905–1915. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Asiedu MK, Beauchamp-Perez FD, Ingle JN,
Behrens MD, Radisky DC and Knutson KL: AXL induces
epithelial-to-mesenchymal transition and regulates the function of
breast cancer stem cells. Oncogene. 33:1316–1324. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Wu X, Liu X, Koul S, Lee CY, Zhang Z and
Halmos B: AXL kinase as a novel target for cancer therapy.
Oncotarget. 5:9546–9563. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Taglienti CA, Wysk M and Davis RJ:
Molecular cloning of the epidermal growth factor-stimulated protein
kinase p56 KKIAMRE. Oncogene. 13:2563–2574. 1996.PubMed/NCBI
|
|
101
|
Gomi H, Sun W, Finch CE, Itohara S,
Yoshimi K and Thompson RF: Learning induces a CDC2-related protein
kinase, KKIAMRE. J Neurosci. 19:9530–9537. 1999.PubMed/NCBI
|