Introduction
Head and neck squamous cell carcinoma (HNSCC) is the
sixth most common type of human cancer with ~750,000 cases
worldwide, which may increase to 1 million by 2030 (1). HNSCC affects subsites of the upper
aerodigestive tract, including the oral cavity, larynx and pharynx,
and the main associated etiological factors are the use of alcohol
and tobacco, and high-risk human papillomavirus (HPV) infection
(2). The tumor suppressor
TP53 is one of the most frequently mutated genes in HNSCC.
Results of The Cancer Genome Atlas (TCGA) HNSC project (https://portal.gdc.cancer.gov/) (3) revealed TP53 somatic mutations
in >90% (357/392) of the cases examined.
HNSCC exhibits heterogeneous tumor phenotypes, as a
result of the reprogramming of the molecular machinery associated
with carcinogenesis. According to Leemans et al (4), at least two genetic subclasses are
identified in HNSCC regarding HPV infection status: i) Tumors with
transcriptionally active HPV that are mostly located in the
oropharynx and generally exhibit wild-type TP53 alleles and
a favorable prognosis; and ii) HPV-negative tumors that often
present with high chromosomal instability, mutated TP53 and
unfavorable prognosis. Low numbers of numerical genetic changes and
wild-type TP53 are also observed in a group of HPV-negative
lesions. Puram et al (5)
demonstrated that the HPV-positive group may likewise exhibit high
levels of chromosomal instability, as well as diversity in HPV
expression, and in cell cycle and senescence states within and
between tumors.
HPV-negative and -positive cases differ with respect
to other characteristics. For instance, alterations in
cyclin-dependent kinase inhibitor 2A (CDKN2A) are frequently
observed in HPV-negative tumors, whereas loss of TNF receptor
associated factor 3 (TRAF3) and amplification of E2F
transcription factor 1 (E2F1) occur frequently in
HPV-positive tumors (6).
CDKN2A (2) and E2F1
(7) have regulatory roles in cell
cycle progression, whereas TRAF3 is involved in the
activation of immune and inflammatory responses (8,9).
Comparing the two groups, the mutational spectrum analyzed by
Seiwert et al (10) also
differs: Mutations in TP53, CDKN2A, cullin 3, discoidin
domain receptor tyrosine kinase 2, F-box and WD repeat domain
containing 7 (FBXW7), lysine methyltransferase 2D/2C
(MLL2/3), nuclear receptor binding SET domain protein 1,
notch receptor 1 (NOTCH1), and
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit α
(PIK3CA); loss of 3p; and amplification of 11q13 and 7p11
potentially targeting cyclin D1, epidermal growth factor receptor
(EGFR) and CDKN2A, respectively, in HPV-negative
cases, and mutations in DEAD-box helicase 3 X-linked, FBXW7,
fibroblast growth factor receptor 2/3, KRAS proto-oncogene GTPase,
NOTCH1 and PIK3CA in HPV-positive cases.
The molecular pathogenesis of HPV-positive HNSCC is
driven through two viral oncoproteins, E6 and E7. E6 and E7 are
overexpressed preceding or just after virus integration (11). E7 triggers the degradation of the
tumor suppressor protein retinoblastoma, releasing E2F and
consequently activating genes that promote the G1-S transition of
the cell cycle. In turn, E6 induces the degradation of the tumor
suppressor p53, a protein that promotes cell cycle arrest,
apoptosis and DNA repair. These events may cause mutations,
interchromosomal rearrangements and synthesis of abnormal
transcripts, explaining the increased cell proliferation and
genomic instability that accelerates the neoplastic process
(12).
In addition to the main role of HPV in the
development of oropharyngeal carcinomas, other viruses, as well as
bacteria and fungi, are related to HNSCC etiology, albeit several
of them with a less direct line of evidence. These examples include
Epstein-Barr virus (EBV) and torque teno virus, which are
associated with nasopharyngeal and laryngeal carcinomas,
respectively. Bacteria and fungi of the oral microbiome appear to
be associated with mouth neoplasms through the production of
carcinogenic metabolites and the conversion of ethanol into
mutagenic and carcinogenic acetaldehyde or promotion of
hypermethylation, proinflammatory events and hypoxic or acidic
environments (13). Such mechanisms
evidence the link between poor dentition or oral hygiene and higher
risks of HNSCC (14).
The initiating events of head and neck tumorigenesis
in HPV-negative lesions are triggered by exposure to alcohol- and
tobacco-derived carcinogens, differing from HPV-positive cases.
Acetaldehyde and tobacco-derived carcinogens (polycyclic aromatic
hydrocarbons and nitrosamines), as well as the resultant
inflammation in exposed tissues, are mainly responsible for these
events. Although the carcinogens are metabolized and excreted,
their metabolites form DNA adducts, which, if not repaired, cause
mutations. Detoxification and DNA repair depend on specific factors
that may be affected by genetic polymorphisms or mutations in genes
involved in carcinogen metabolism, as observed in Fanconi anemia, a
rare genetic disease caused by a deficiency in DNA repair
mechanisms, with an increased risk of developing HNSCC (6).
Conventional primary treatments for HNSCC are lesion
resection and radiotherapy for early stages of the disease, and
chemotherapy for locally advanced disease. Biological and
chemotherapeutic agents combined with radiotherapy have
demonstrated high levels of efficiency in local control and patient
survival (15). For patients with
recurrent or metastatic tumors, therapeutic options include immune
checkpoint inhibitors, platinum derivatives, fluorouracil (FU) and
cetuximab (CTX) (16). CTX is a
monoclonal antibody that competitively targets the extracellular
domain of EGFR, blocking proliferative, antiapoptotic and
proangiogenic signals (17) Other
EGFR inhibitors have been developed; however, they have not
significantly improved the rates of patient survival, and are
associated with high levels of toxicity (18).
Chemotherapy is one of the most commonly used
treatments. However, drug resistance is a major obstacle in
controlling disease progression. Tumors may be nonresponsive to a
particular treatment due to intrinsic (or primary) resistance
caused by inherited mutations or insensitive clones, whereas other
tumors develop resistance (acquired or adaptative) after a positive
response to therapy (14). Cells
that survive initial chemotherapy represent the population
reservoir from which resistant clones may emerge (15).
Several factors contribute to the development of
drug resistance and understanding the mechanisms involved is
crucial to improve cancer treatment. They include increased drug
efflux, reduced drug uptake, drug inactivation, resistance to
apoptosis, efficient DNA repair, epigenetic changes that affect
gene expression, drug target mutations, overexpression or
amplification of genes associated with resistance and genomic
instability (19). Underlying these
mechanisms are specific biological processes that are critical to
drug resistance. For instance, selective protein degradation was
shown to regulate the expression of three ATP-binding cassette
transporters that limit drug uptake by cells (20). Evasion of apoptosis may equally be
regulated by protein degradation and noncoding RNA or DNA
demethylation through the downregulation of proapoptotic proteins
(21,22).
Extrinsic factors, such as tumor microenvironment
components, can also promote resistance. Su et al (23) showed that a carcinoma-associated
fibroblast subpopulation that exhibits two cell-surface markers,
membrane metalloendopeptidase (neprilysin) and complement C5a
receptor 2 (C5AR2), provides a survival niche for cancer stem
cells, which are tumorigenic and chemoresistant in certain cancer
types. In addition, neprilysin+C5AR2+
fibroblasts are resistant to chemotherapy and can induce
chemoresistance in tumor cells by secreting interleukins IL-6 and
IL-8.
Thus, chemotherapy resistance poses a complex
challenge in cancer treatment. The present review aimed to address
the potential relationship between tumor evolution,
epithelial-mesenchymal transition (EMT), EGFR signaling alterations
and drug resistance in HNSCC, with a focus on CTX chemotherapy.
Tumor evolution
Regarding the clinical evolution of HNSCC tumors,
initial oral cavity lesions present as a painful mass or ulcer that
compromises eating or speaking. Oropharyngeal and hypopharyngeal
tumors are usually diagnosed at later stages with symptoms of
dysphagia, odynophagia or otalgia, and HPV-positive cases may
remain asymptomatic for numerous years. Early laryngeal
manifestations include change in voice or hoarseness, but after
evolving to a more advanced stage, they may compromise airway
patency, leading to dyspnea. Clinical manifestations of
nasopharyngeal tumors with EBV infection are stage-dependent, and
initial cases may present with a solitary, painless cervical mass
and unilateral nasal obstruction, which evolve to display other
more severe symptoms, including otalgia, epistaxis, vision changes,
headache and persistent rhinorrhea (6,24).
The evolution of the tumor mass is gradual. For
numerous years, the progression of genetically transformed cells to
a malignant condition was considered an isolated process. During
this process, early neoplastic lesions recruit and activate stromal
cells, such as fibroblasts, pericytes and adipocytes, as well as
immune cells, to promote a microenvironment favorable to disease
progression. These cells then alter the tumor microenvironment
through the secretion of factors that maintain proliferative
signaling and activate invasion and metastasis, ultimately favoring
tumor cell homeostasis (25,26).
In addition to biological agents, physical factors may alter the
extracellular matrix, cytoskeleton and blood vessel permeability.
These factors may also affect genome integrity and gene expression
or localization of effectors that control the cell cycle, such as
CDKN1B, NF-κB p105 subunit and transcriptional coactivator YAP1/WW
domain-containing transcription regulator protein 1, contributing
to neoplastic transformation, EMT and tumor evolution from abnormal
growth patterns to increased tissue volume (27,28).
Regarding neoplastic cells, malignant transformation
occurs through successive mutations that result in acquired
capabilities or hallmarks. Hanahan and Weinberg (29) defined the hallmarks of cancer, which
include sustained proliferative signaling, evasion of growth
suppressors, resistance to cell death, replicative immortality,
sustained angiogenesis, invasion and metastasis. A decade later,
Hanahan and Weinberg (30) proposed
two additional interrelated emerging hallmarks: Reprogramming of
energy metabolism and evasion of immune destruction, which depend
on genome instability and tumor-promoting inflammation, like other
hallmarks. Recently, Hanahan (31)
detailed the addition of two new hallmarks-unlocking phenotypic
plasticity and nonmutational epigenetic reprogramming- and two
enabling characteristics or ‘facilitators’ for the acquisition of
hallmarks-senescent cells and polymorphic microbiomes.
Underlying the hallmarks are driver mutations, which
are genomic alterations that confer growth or a survival advantage
to the cell. Unlike driver mutations, passenger mutations have no
role in the malignant phenotype and may be lost during the
neoplastic process, but contribute to the tumor mutational burden,
which is a potential biomarker of response to therapy (32).
Bypassing cell cycle checkpoints and increasing
proliferation are achieved by synthetizing growth factors or
receptors and inducing neighboring cells to produce ligands of
interest. A similar effect is also attained through structural
changes and amplifications of receptor molecules or through
downstream effectors that maintain signals without requiring
external stimuli (26). Further
mutations modify the tumor microenvironment and allow tumor cells
to pass through the extracellular matrix and reach other tissues.
The overexpression of cytokines and growth factors promotes
inflammatory infiltration and growth of blood and lymphatic
vessels. The acquisition of such characteristics occurs through
gene mutations and epigenetic modifications, as well as Darwinian
selection, which drives the evolution of a healthy cell to a
malignant population, allowing adaptation to new environmental
pressures and progression to novel phenotypes (33).
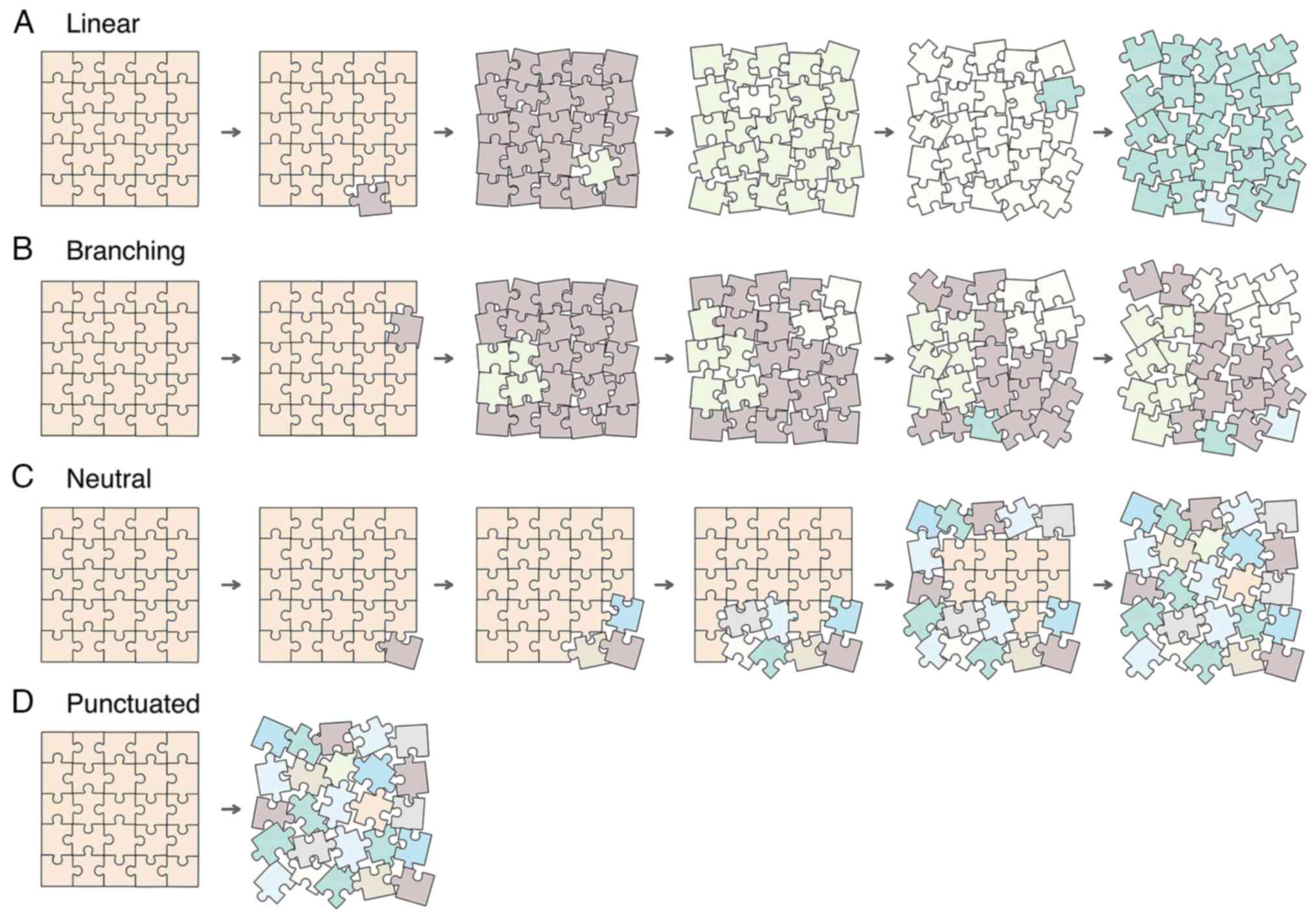
Nowell (34) stated
that cancer arises from a single cell and evolves linearly through
the selection of mutations, resulting in a homogeneous clone with a
strong selective advantage (Fig.
1A). Dexter et al (35)
and others (36,37) have proposed a model in which tumors
evolve in a branched fashion. According to this model, distinct
mutated clones derived from the same clone are differentially
selected by endogenous and external factors, which results in high
intratumor heterogeneity (Fig. 1B).
Conversely, Williams et al (38) proposed that alterations responsible
for the neoplastic process are present in the first malignant cell
and the subsequent mutations are neutral (Fig. 1C). Results of Williams et al
(38) revealed that carcinomas of
the stomach, lung and cervix display neutral evolution; pancreatic
and thyroid carcinoma and glioblastoma display profiles compatible
with nonneutral evolution, and HNSCC displays mixed evolution
(Table I).
 | Table I.Models of Cancer Evolution: Linear,
branching, neutral and punctuated (refs. 33–48). |
Table I.
Models of Cancer Evolution: Linear,
branching, neutral and punctuated (refs. 33–48).
| Evolution type | Mechanism of
evolution | Intratumor
heterogeneity | Examples |
|---|
| Linear | Genetic alterations
successively selected step-by-step resulting in a major dominant
clone | Low | Acute lymphoblastic
leukemia |
| Branching | Co-existence and
continued evolution of multiple subclones derived from a common
ancestor | Variable | Breast, liver,
colorectal and prostate cancers |
| Neutral | Acquisition of
driver followed by random fixation of neutral mutations through
genetic drift | High | Stomach, lung and
cervical carcinomas |
| Punctuated | Gradual acquisition
of driver mutations interspersed with rapid clonal expansion | High | Prostate
cancer |
| Mixed | Shift from one to
another evolution type or multiple types simultaneously | High | Head and neck
carcinoma |
In nonneutral evolution, continuous clonal selection
and adaptation to microenvironment niches have important roles in
tumor progression. Groups of premalignant cells that undergo clonal
expansion following chronic exposure to environmental carcinogens
exhibit a high risk of generating multiple local primary tumors, a
process that has been reported in various cancer types, including
HNSCC. This process is known as field cancerization and may provide
an explanation for the presence of tissue areas positive for p53 on
immunostaining, prone to malignant transformation and with high
rates of recurrence (39). In
addition, studies on yeast or bacteria have revealed that neutral
synonymous mutations in protein-coding genes, which do not change
protein sequences, may affect mRNA structure, level or function and
alter growth rates, particularly under strong selection (40,41).
Therefore, although Williams et al (38) and Li et al (42) have shown that a significant number
of neoplastic subclones undergo neutral evolution, new biological
or physical selective pressures, such as therapy, extracellular
matrix remodeling or tissue structure, may promote the expansion of
neutral clones, resulting in EMT, metastasis and tumor progression
(28).
Studies on cancer biology have identified various
single-nucleotide mutations and small insertion or deletion driver
mutations that may be acquired over time. However, several tumor
types exhibit complex chromosome aberrations that involve localized
(chromothripsis) or dispersed (chromoplexy) genomic regions
(43–46), which define a punctuated type of
evolution (33) (Fig. 1D; Table
I). These catastrophic events occur early in the neoplastic
process, resulting in a rapid ‘big bang’ clonal expansion, which is
characterized by uncontrolled cell proliferation (47,48).
This instability is often unfavorable and may halt or even reverse
tumor growth through selective pressures intrinsic to cellular
metabolism or part of the immune response, as well as through
factors from the tumor microenvironment (30). However, if successful, instability
may lead to the development of aberrant cells that drive metastasis
and therapeutic resistance (49,50).
By combining the evolutionary view of cancer with
oncogene and tumor suppressor gene concepts, Fearon and Vogelstein
(51) proposed a colorectal
carcinogenesis model in 1990, in which mutations build linearly
step-by-step. This sequence of events was later investigated in
numerous other cancer types. Recent pan-cancer next-generation
sequencing data have provided insight into intratumoral
heterogeneity, which has a role in multiple shared evolutionary
trajectories associated with prognostic biomarkers (52). In certain cases, convergent
evolution is observed, with distinct mutations acquired in the same
gene or pathway (49,53,54).
By contrast, other tumors show no evidence of subclonal driver
mutation enrichment, but exhibit passenger mutations, which are not
associated with cell growth and survival, but may have a role in
intratumoral heterogeneity (49).
Martínez-Jiménez et al (55)
reported that metastatic lesions exhibit low intratumoral
heterogeneity, which is consistent with the findings of Nguyen
et al (56). These results
were observed in numerous tumor types, including head and neck
carcinomas, and may be explained by a dominant clone in the seeding
event or selective external pressure.
Dynamic evolution associated with resistance to
chemotherapy occurs in parallel to cancer evolution, generating
adaptive costs for neoplastic cells in the form of vulnerability
windows, known as persistent or temporary collateral sensitivity
(57). This condition is also
demonstrated in Plasmodium and bacteria and is based on the
observation that a trait, such as drug resistance, may occur in
detriment to another trait (58).
The vulnerability window presents opportunities for new therapeutic
approaches, combining the identification of tumor propensity toward
resistance and sensitivity during tumor evolution.
These findings highlight the clinical relevance of
tumor evolution model studies, as the mechanisms discussed may
contribute to further understanding the complexities of cancer
treatment. Therefore, identifying the driver markers of aggressive
clones that proliferate under therapeutic and biological or
physical pressures is important because they can predict resistance
(59). It is important to identify
subsequent metabolic reprogramming that supports a stress condition
capable of selecting new mutations responsible for conferring
higher phenotypic variability and adaptation (60).
In addition to genetic mechanisms, including
mutations and chromosome aberrations, cancer cells can change from
one phenotype to another through epigenetic and transcriptional
adaptive processes without genomic alterations. These nongenetic
mechanisms of cancer evolution may culminate in
transdifferentiation to a dissimilar subclone and reversion to a
progenitor phenotype or EMT, and represent a new challenge in the
prediction and treatment of cancer resistance (61).
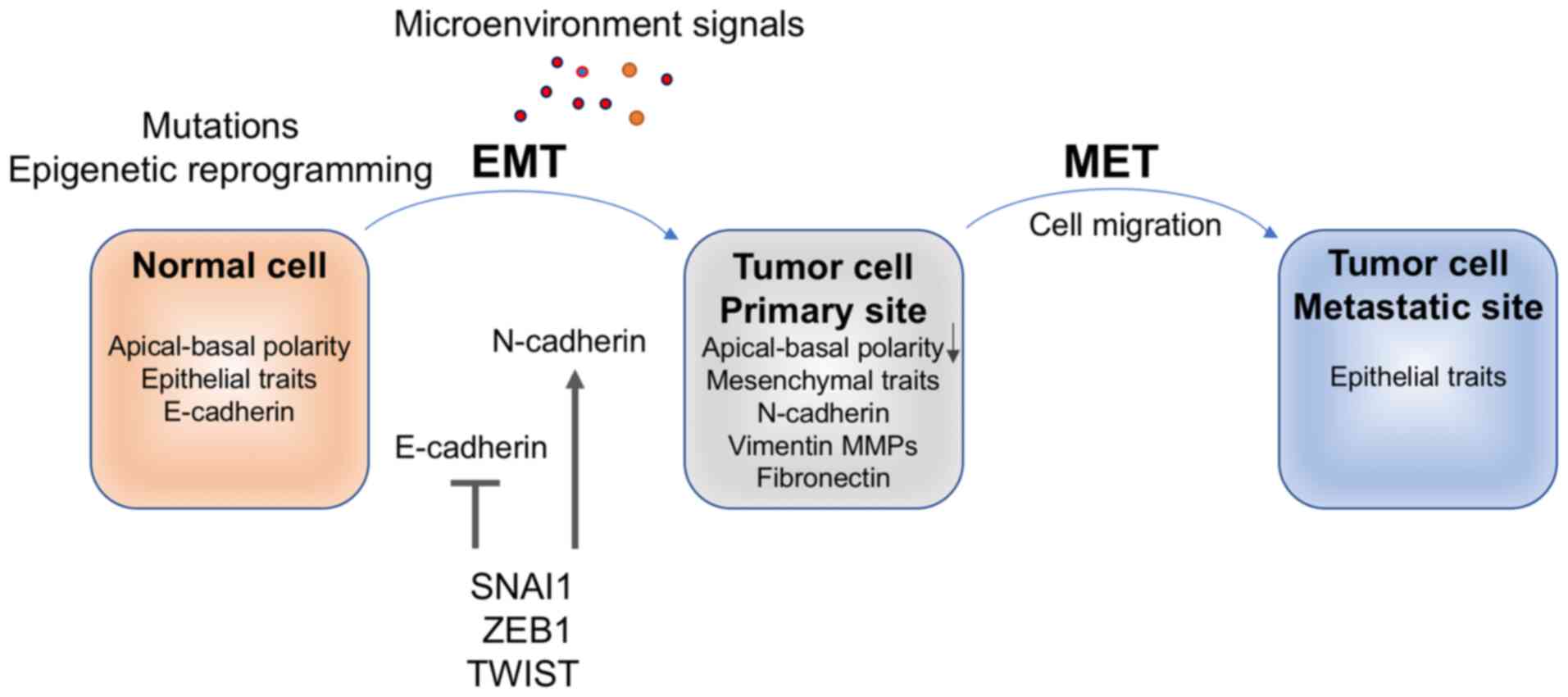
EMT
EMT is a cellular differentiation program and a
phenotypic shift that culminates in the dissolution of epithelial
cell-cell-contacts, such as desmosomes, tight junctions, adherens
junctions and gap junctions, through the disruption of the Crumbs,
partitioning-defective (PARD) and Scribble polarity complexes.
Consequently, cells lose apical-basal polarity and epithelial
traits (62). Expression of the
epithelial cell adhesion protein E-cadherin is downregulated and
expression of mesenchymal proteins, including N-cadherin, vimentin
and fibronectin, is activated (63,64).
The subsequent reorganization of the epithelial actin
microfilaments allows for motility through membrane projection,
actin contraction and adhesion.
EMT is required for morphogenesis during embryonic
development. The resulting mesenchymal lineage exhibits migratory
properties that allow them to be recruited to specific sites in the
embryo, where they undergo mesenchymal-epithelial transition (MET)
and form new epithelial tissues (65). When aberrantly activated, EMT and
MET contribute to neoplastic progression by allowing cells to
migrate to other organs through lymphatic or hematogenous
dissemination and metastasize. Due to intratumoral heterogeneity,
different levels of EMT activation occur according to the area of
the tumor mass, resulting in partial EMT (p-EMT) or total EMT
(66). This may be explained by
nonmutational epigenetic plasticity. In oral squamous cell
carcinomas, the invasive front demonstrates p-EMT, with a loss of
transcription factors directly involved in EMT, and the expression
of other EMT-defining genes, which are absent in the central core
of the tumors. This heterogeneity has not been associated with the
presence of mutations in different tissue areas and may be
attributed to epigenomic variability resulting from histone
modification, DNA methylation and posttranscriptional modification
of RNA (31). However, paracrine
p-EMT regulation by microenvironmental stroma cannot be excluded
(67).
Results of in vitro studies have demonstrated
that cell lines may display different rates of p-EMT states and
varying EMT phenotypes. Many exhibited a more epithelial phenotype,
whereas others appeared to be mesenchymal with a higher migration
rate (68). Cell migration consumes
high levels of ATP and mesenchymal phenotypes may increase aerobic
glycolysis. DeCamp et al (69) and others (70,71)
focused on a specific process that promotes the acquisition of a
plastic phenotype by epithelial cells, allowing them to perform
collective epithelial-cell migration through mechanisms other than
EMT, known as epithelial unjamming. This leads to a fluid-like
migratory phase and a shift toward glycolytic energy metabolism
without cell-cell junction disruption. It involves lipid, cellular
ketone and carbohydrate metabolism, with the oxidation of fatty
acids for ATP or energy generation in mitochondria. The authors
hypothesized that epithelial unjamming may be an adaptation that
allows the confluent epithelial collective to perform dynamic
events, such as those occurring during embryonic development, wound
healing and neoplastic progression, at the cost of high energy
expenditure.
EMT is associated with stimuli in the tumor
microenvironment and signaling pathways, including Wnt/β-catenin,
NOTCH, transforming growth factor (TGF)-β/SMAD, PI3K/AKT and
JAK/STAT, in which ligands and receptors, such as IL-6 (72,73),
TNF (74), TGF, bone morphogenetic
proteins (62,75) and tyrosine-kinase receptors
(75), interact. These signaling
pathways regulate the expression of transcription factors and their
epithelial targets, such as E-cadherin, and mesenchymal targets,
such as N-cadherin, vimentin, fibronectin and
MMPs/metalloproteinases. Numerous genes are associated with EMT.
The EMT gene database (dbEMT; http://dbemt.bioinfo-minzhao.org/; version 2.0) lists
1,184 EMT-related genes, 1,011 protein-coding genes, and 173
noncoding RNAs (57). However, few
genes are directly associated with EMT, such as the transcription
regulators zinc finger protein SNAI1 (SNAI1), zinc finger
E-box-binding homeobox 1 (ZEB1), ZEB2 and twist family bHLH
transcription factor (TWIST), which bind to specific DNA sequences
in promoters or enhancers through zinc finger domains, homeodomains
or helix-loop-helix domains that activate the transcription of
target genes (76).
SNAI1 and ZEB1 induce EMT by binding to the promoter
of the E-cadherin gene (cadherin 1; CDH1) and the promoters
of MMP2 and MMP9, repressing and promoting their
transcription, respectively. ZEB1 also stimulates the expression of
SETD1B (histone-lysine N-methyltransferase SETD1B), a
histone methyltransferase that influences chromatin organization,
in addition to a positive feedback loop with ZEB1. Similarly,
upregulation of SNAI1 causes alterations in the chromatin state,
which may consequently activate EMT genes. TWIST modulates
mesenchymal and epithelial phenotypes, thus regulating the
transcription of both N-cadherin/CDH2 and
E-cadherin/CDH1, respectively (76) (Fig.
2).
The contribution of SNAI1, ZEBs and TWIST to EMT is
subordinated to the cell type and the presence of activating
ligands and stimuli in the tumor microenvironment (75). For instance, Puram et al
(67) did not detect the expression
of classical EMT transcription factors in an analysis of the HNSCC
single-cell transcriptome. However, results of the present study
demonstrated that most epithelial and mesenchymal EMT markers,
including vimentin, integrin, TGF-β-induced genes and SNAI2, were
maintained. Thus, Puram et al (67) hypothesized that the p-EMT state
localized at the leading edge of the tumor is distinct from the
full EMT state and may be due to paracrine interactions between
stromal and neoplastic cells. Puram et al (67) also determined the potential
association between high p-EMT and number of lymph node metastases,
tumor grade and adverse pathological characteristics, including
extracapsular extension and lymphovascular invasion. Of note,
overexpression of factors involved in EMT has been associated with
metastasis, advanced stages of disease and low overall survival
rates in patients with various cancer types (76,77),
and were confirmed in HNSCC (78,79).
Okuyama et al (80)
highlighted the association of tumor budding (defined as the
presence of isolated clusters of up to five cancer cells ahead of
the invasive tumor front) with p-EMT, and determined the potential
of tumor budding as a prognostic marker for poor survival in HNSCC.
Therefore, these biomarkers are potential targets for cancer
treatment.
EGFR and HNSCC
Receptor tyrosine kinases (RTKs) are key regulators
of EMT. EGFR is an RTK that has an important role in cell
physiology by regulating several signaling cascades involved in
critical cellular functions such as proliferation, differentiation,
survival and motility. However, when EGFR is mutated or
overexpressed, it can trigger a neoplastic process (81).
EGFR is a transmembrane glycoprotein and a member of
the ErbB tyrosine-kinase family encoded by EGFR/ERBB1/HER1
at 7p11.2 [gene ID, 1956; National Center for Biotechnology
Information (NCBI), https://www.ncbi.nlm.nih.gov/gene/1956]. EGFR
possesses an extracellular ligand-binding region and a
cytoplasmatic kinase domain connected by a transmembrane helix. The
extracellular region of EGFR has four domains: I and III are
leucine-rich domains for ligand binding, while II and IV are
cysteine-rich domains. Binding of the EGFR ligands, such as EGF,
amphiregulin, betacellulin, epigen, epiregulin heparin-binding EGF
and TGF-α (82) promotes a
conformational change from a self-inhibited ‘tethered’ state to an
open ‘untethered’ state. The new conformation facilitates domains
II and IV to bind to an adjacent RTK and homo- or heterodimerize,
inducing auto-phosphorylation, activation and recruitment of
effectors to initiate a signaling cascade (83,84).
Purba et al (85) proposed
that EGFR is an inactive dimer prior to ligand-induced
dimerization. When the ligand binds the receptor, its transmembrane
sequence may undergo a rotation, resulting in an active
configuration of the intracellular domain of the RTK. Hence,
mutations in the extracellular sequence may promote the rotation
and activation of the receptor without the ligand (84) (Fig.
3).
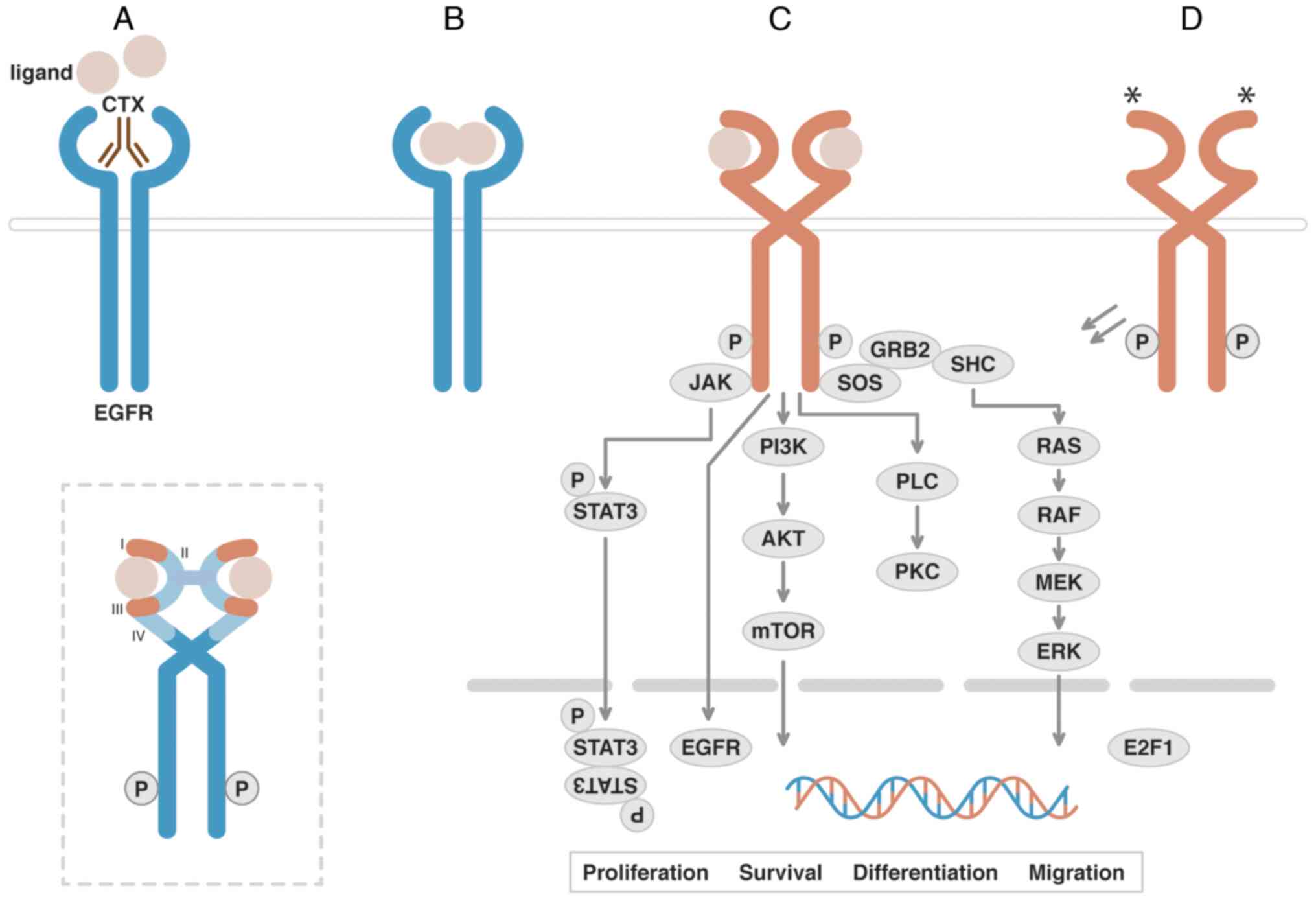 | Figure 3.EGFR Signaling Transduction and
Downstream Effectors. (A) EGFR is a receptor tyrosine kinase that
possesses an extracellular ligand-binding region and a
cytoplasmatic kinase domain connected by a transmembrane domain.
CTX is a chimeric human/mouse monoclonal antibody that binds to
domain III of the receptor blocking binding of natural ligands. (B)
Binding of the EGFR ligands promotes a conformational change that
facilitates homo- or heterodimerization, inducing (C)
autophosphorylation, activation and recruitment of effectors to
initiate a signaling cascade. (D) Mutations (*) in the
extracellular sequence may promote the activation of the receptor
without the presence of a ligand. The dashed box highlights a
schematic diagram of extracellular ligand-binding domains (I and
III) of EGFR. AKT, RAC serine/threonine-protein kinases; E2F1,
transcription factor E2F1; EGFR, epidermal growth factor receptor;
ERK, extracellular signal-regulated kinases; GRB2, growth factor
receptor-bound protein 2; JAK, Janus kinase; MEK, MAP kinase
kinases; mTOR, mammalian target of rapamycin; PI3K,
phosphoinositide 3-kinase-C2-α; PKC, protein kinase C; PLC,
phospholipase C-γ-1; RAF, proto-oncogene c-RAF; RAS,
GTPase-activating protein; SHC, SHC-transforming protein 1; SOS,
son of sevenless homolog 1; STAT3, signal transducer and activator
of transcription 3; I, II, III, IV, EGFR extracellular domains. |
In 232 HNSCC cases analyzed in the TCGA HNSC
project, 17 (7.33%) somatic mutations and no copy number variation
gains or losses were observed in EGFR. However, upregulation
of EGFR and associated ligands is frequent in HNSC (6,86),
which may be a result of the expression of inflammatory mediators
and transcription factors caused by external factors, such as
tobacco smoke. G protein-coupled receptor signaling (82) activation by p53 protein,
polymorphisms in intron 1 of EGFR, and EGFR
amplification (85,87) are alternate mechanisms that increase
EGFR expression or synthesis of associated ligands. Kriegs
et al (86) demonstrated
that EGFR expression and autophosphorylation in HNSCC cells are not
well correlated, and may be dependent on other factors, such as
EGFR polymorphisms and mutations in downstream effectors.
Pleiotropic functions of EGFR in HNSCC and other
tumors are associated with MAPK/ERK (proliferation,
immunosuppression, angiogenesis) (88,89),
PI3K/AKT (cell survival and proliferation; apoptosis evasion)
(89,90), JAK/STAT (cell growth, development,
differentiation and survival) (91)
and phospholipase (PLC) C-γ-1/protein kinase C (PKC)
(proliferation, migration) (92,93).
Particularly when mutated or overexpressed in HNSCC, EGFR has
protumorigenic and prometastatic roles associated with nutrient
uptake and biosynthesis, proliferation, inflammation, cell
survival, migration and invasion. In addition to the membrane-bound
functions, EGFR activation may result in its endocytosis and
translocation to the nucleoplasm, where it modulates the activity
of numerous genes, including those associated with the cell cycle,
such as CCND1, DNA damage response, e.g. the catalytic
subunit of the DNA-dependent protein kinase, DNA replication (e.g.
proliferating cell nuclear antigen, cofactor of DNA polymerase),
mitochondrial electron transport, such as cytochrome c oxidase
subunit II, and inflammation, e.g. nitric oxide synthase 2
(94).
Multiple strategies have been developed to inhibit
EGFR in HNSCC, such as monoclonal antibodies that compete for the
ligand-binding region to prevent the activation of the
cytoplasmatic tyrosine kinase domain and small inhibitor molecules
that bind to the kinase domain to block EGFR autophosphorylation
and downstream signaling. CTX, zalutumumab, panitumumab and
nimotuzumab are examples of anti-EGFR antibodies, and gefitinib,
erlotinib and lapatinib are tyrosine kinase inhibitors. CTX is
approved by the Food and Drug Administration as an anti-EGFR agent
for use in HNSCC chemotherapy. Several studies have detailed the
associated efficacy and adverse events (95–97).
CTX therapy, and head and neck
carcinoma
The majority of head and neck carcinoma cases are
diagnosed at late stages and thus associated with short
progression-free survival. Current treatment options aim to improve
overall survival and delay disease progression while maintaining
quality of life. The current conventional treatment involves
surgical intervention and lesion irradiation for early-stage cases
and chemotherapy for advanced cases. The results of a meta-analysis
involving 107 randomized trials and 19,805 patients demonstrated
that adjuvant chemotherapy following primary treatment involving
surgical lesion removal or radiotherapy did not increase overall
survival. Furthermore, certain levels of toxicity were observed. By
contrast, concomitant chemotherapy for locally advanced disease
exerted positive effects on overall survival. However, in patients
aged ≥70 years, overall survival rates were markedly decreased
following concomitant chemotherapy; thus, treatment options should
be discussed thoroughly with multidisciplinary health teams and
patients' families (98).
Platinum derivatives with FU + CTX are considered
the standard of care chemotherapy treatment for recurrent or
metastatic HNSCC. This treatment option (EXTREME protocol)
significantly increased the median overall survival of patients
from ~7 months in the chemotherapy-alone group to ~10 months in the
group that received chemotherapy plus cetuximab (99–101).
Cisplatin is an inorganic platinum derivative that induces DNA
intrastrand crosslinks, subsequently interfering with DNA
replication and transcription, leading to cell-cycle arrest and
apoptosis (102). FU, a
fluorinated pyrimidine analogue, causes cellular cytotoxicity
through a complex metabolic process. For instance,
fluorodeoxyuridine triphosphate can be incorporated into nucleic
acids affecting DNA and RNA functions, whereas the metabolite
fluorodeoxyuridine monophosphate, resulting from the conversion of
FU by thymidine phosphorylase and thymidine kinase, forms a stable
complex with thymidylate synthase, resulting in thymidine
depletion, decreased DNA synthesis and cell lethality (103).
CTX is a chimeric human/mouse immunoglobulin
G1-subclass monoclonal antibody drug that binds to the
extracellular ligand-binding region (domain III) of EGFR (Fig. 3A) with higher affinity than the
natural ligands EGF and TGFα. CTX blocks the untethering
conformation of the receptor monomer, further inhibiting
dimerization, eventually halting the activation of the tyrosine
kinase domain and proliferation signaling through RAS and ERK
(104,105).
Other antitumor mechanisms of CTX are inhibition of
ligand-receptor binding, internalization and degradation of EGFR,
and antibody-mediated cytotoxicity (106). CTX also activates proinflammatory
and proapoptotic factors and inhibits repair of radiation-induced
lesions, increasing the efficacy of strategies that combine
EGFR-targeted agents and immune- or radiotherapy. CTX blocks
invasion, angiogenesis and metastasis, which are protumorigenic
roles of several signaling pathways associated with EGFR (81,107).
By contrast, activating mutations in members of the RAS pathway may
reduce the efficacy of CTX, leading to an improper ERK response and
changes in transcription, cell fates and proliferation (108).
Jie et al (109) observed that in HNSCC, CTX therapy
increased intratumoral regulatory T cells with an immunosuppressive
phenotype, but promoted impaired expression of molecules related to
antibody-dependent cellular cytotoxicity of natural killer cells in
the tumor microenvironment. This inverse correlation indicates that
regulatory T cells restrain infiltrating natural killer
cell-mediated cytotoxicity, which negatively affects CTX therapy in
this group of tumors. CTX also exerts synergistic effects, such as
increasing radiation-induced apoptosis by blocking DNA repair
mechanisms dependent on PI3K/AKT, MAPK/ERK and JAK/STAT pathways
(104).
Different mechanisms of action of CTX were also
observed in HNSCC, such as upregulation of the transcription factor
encoded by transcription factor AP-2α (TFAP2A) by Kagohara
et al (110). Their results
suggested that TFAP2A induces cell proliferation and is
potentially overexpressed by CTX to overcome EGFR inactivation. The
authors also detected upregulation of the AXL gene, which
encodes a receptor tyrosine kinase related to growth, migration and
inflammation, and overexpression of several EMT genes of
collagenases.
As CTX is a large monoclonal antibody, it cannot be
filtered by the kidneys. Thus, a small fraction is eliminated by
biliary excretion, and the majority is eliminated through
intracellular catabolism (111).
CTX may be administered alone in cases eligible for radiotherapy
when platinum-based therapy is not appropriate (112). Contraindications to receiving
cisplatin consider factors such as the Eastern Cooperative Oncology
Group Performance Status score, age, comorbidities, involuntary
weight loss, concomitant medications and prior platinum-based
chemotherapy (112–115).
Alternate regimens were developed to enhance the
overall and progression-free survival rates in patients with HNSCC.
For instance, the TPEx protocol, which combines the taxane
docetaxel, CTX and cisplatin, increased median overall survival to
14 months (116) and improved
tolerance to treatment and quality of life (100), compared with the EXTREME protocol.
In patients with HPV and oropharyngeal cancer, results of the
De-ESCALaTE HPV trial demonstrated that CTX exerted no benefits in
toxicity or tumor control compared with radiotherapy plus cisplatin
(117). This result is expected,
because HPV-positive cases of HNSCC are associated with the viral
oncoproteins E6 and E7 rather than the EGFR protumorigenic
signaling pathways targeted by CTX (118).
Primary or intrinsic resistance to CTX may occur in
a small number of patients with HNSCC; however, almost all patients
develop acquired resistance. Intrinsic mechanisms of resistance
include alterations in EGFR and the associated ligands or
effectors, acquired mutations in genes of alternative oncogenic
pathways involved in tumorigenesis, such as Ras/Raf/MAPK/ERK and
PI3K/AKT/mTOR, loss of phosphatase and tensin homolog (PTEN) or
phosphorylation of STAT3, and epigenetic modifications. In contrast
to intrinsic resistance, mechanisms associated with acquired
resistance in HNSCC are diverse and include alterations in EGFR and
its ligands, activation of the PI3K/AKT/mTOR signaling pathway,
loss of PTEN, EMT phenotype acquisition, phosphorylation of STAT3,
epigenetic alterations and an immunosuppressive tumor
microenvironment (104,119–122). For example, EGFR mutations in
subdomain I or close to the EGF-binding pocket (G33S and N56K)
promote an open untethered receptor with a reduced affinity for EGF
and CTX, but with decreased degradation and sustained activation of
AKT signaling (84). The literature
on CTX resistance in HNSCC has also reported abnormal expression of
markers directly associated with EMT, including vimentin (110), ZEB2, TWIST1, SNAIL, E-cadherin and
fibronectin, and markers related to EMT process, such as MAD
homolog 4, main components of the epithelial cytoskeleton (keratins
13, 14 and 16), and the metalloproteinase ADAM 19 (121,123,124), as well as alternate mechanisms of
resistance or sensitivity associated with microRNA-9 (125) and polymorphisms of cytochrome P450
1B1 (126).
Using high-throughput screening to examine the
activity of 42 RTKs, Wheeler et al (127) reported that EGFR, HER2 and HER3
are highly activated in CTX-resistant HNSCC cells. The mechanisms
involved comprise a dysregulation of EGFR internalization or
degradation and EGFR-dependent activation of HER2 and HER3, which
initiate a proliferative and survival signaling cascade.
In addition to the HER family and the downstream
effectors, EGFR blockade in HNSCC results in the activation of
several alternative growth factor receptor pathways, such as
anaplastic lymphoma kinase (ALK), insulin-like growth factor
(IGF)-1 and hepatocyte growth factor (MET), which are members of
the RTK family (128). MET
activation modulates several signaling cascades, including
PI3K/AKT, JAK/STAT, Ras/MAPK, SRC and Wnt/β-catenin (129). MET has been found to be increased
in lymph node metastases of HNSCC, compared with primary tumors
(130). IGF-1 receptor (IGF-1R) is
associated with tumorigenesis of epithelial cancers. Its activation
potentially requires EGFR kinase activity or IGF-1R/EGFR complex
formation, and stimulates G1- to S-phase transition in a PI3K/AKT
and ERK-dependent pathway (131).
ALK is a marker upregulated in advanced HNSCC, compared with
early-stage tumors (132).
Functional assays and evaluation of receptor expression or
activation and mutational status of effectors indicate that MET and
ALK activation, as well as increased heterodimerization of EGFR and
IGF-1R, are mechanisms of CTX resistance in HNSCC (133–135).
Umemori et al (136) observed increased expression of
epithelial cell adhesion molecule (Ep-CAM) products in HNSCC
samples. Ep-CAM is a transmembrane glycoprotein that promotes cell
adhesion, cell proliferation, EMT and cancer stemness, and is a
potential prognostic marker for human carcinomas. Its N-terminus
(EpEX) exhibits EGF-like domains that can function as ligands for
EGFR (137,138). Umemori et al (136) showed that EpEX competes against
CTX and stimulates the EGFR-ERK pathway, contributing to
resistance.
Furthermore, an energetic metabolic shift during
treatment has been observed. Results of a study using HNSCC
patient-derived tumor xenografts revealed that the lactate to
pyruvate ratio was significantly decreased in CTX-sensitive
xenografts between pretreatment and posttreatment, but it was not
affected in CTX-resistant xenografts (139). These results indicate that a
glycolytic phenotype may contribute to the development of CTX
resistance.
Several studies have suggested that resistance to
anti-EGFR therapies is related to hypoxia and angiogenesis. Hypoxia
occurs in conditions with high demands of oxygen due to increased
proliferation rates and deficient angiogenesis These conditions
induce the expression of hypoxia-inducible transcription factors
(HIFs), which are responsible for upregulating genes involved in
oxygen supply (e.g., in modulating angiogenesis) and genes that
limit oxygen consumption (involved in nonoxidative metabolism). In
tumor cells, HIFs also activate genes that promote EMT, immune
evasion, reprogramming of energy metabolism and acquisition of
cancer stem cell properties. Vascular endothelial growth factor and
other angiogenic growth factors, such as stromal-derived factor 1,
stem cell factor and angiopoietin family members, are also
regulated by HIFs (140). As
expected, EGFR pathway inhibition decreases HIF and vascular
endothelial growth factor expression and renders the tumor
sensitive to antiangiogenic therapies; thus, combining the two
approaches may improve outcomes (141,142).
In patients with HNSCC, hypoxic tumors show a
microenvironment with increased numbers of immunosuppressive
regulatory T cells, a feature that may contribute to a deficient
immunotherapy response. These tumors also show enrichment of
hypoxia and EGFR and TGF-β signaling genes and decreased expression
of interferon (IFN)α and IFNg effectors. A significant shift toward
hypoxia normalization and growth of T- and B-cell subsets was
observed after CTX therapy prior to surgery. These findings
indicate that CTX combined with immunotherapy may overcome
resistance in a group of patients with hypoxic tumors (107). An in vitro study
demonstrated that HNSCC cell lines exhibit sensitivity to CTX under
hypoxic conditions, inducing downregulation of HIF-1α and reduced
growth. However, both HIF-1α suppression and CTX treatment were
unable to inhibit EMT and reduce the expression of stem cell
markers. Therefore, at least in vitro, resistance to CTX in
HNSCC cells appears to be independent of hypoxia conditions
(143).
Ge et al (144) demonstrated that in patients with
HNSCC treated with CTX, dynamic changes in the levels of T-cell
receptors in peripheral blood and tumor tissue occur, which may be
associated with therapeutic responses. These results highlight the
importance of evaluating T-cell receptors and determining their
potential use as a noninvasive approach for assessing response to
CTX in HNSCC. In addition, monitoring clonal composition and
circulating molecules, while analyzing three-dimensional systems,
such as patient-derived organoids and organotypic culture, may aid
in understanding tumor progression and therapeutic resistance
mechanisms (104,122,145).
Resistance to therapy is a major task in cancer
treatment and occurs through numerous routes: Drug targets acquire
mutations or are downregulated; alternative pathways are activated;
drug pumps are upregulated; xenobiotic receptors, detoxicating
enzymes and efflux transporters are downregulated; metabolic
profiles of the tumor and its microenvironment are changed;
apoptosis resistance and immune evasion are stimulated; and DNA
repair is altered. Extrinsic factors include hypoxia, inflammation,
immune response, and other microenvironment characteristics. Many
solutions for this scenario can be proposed, such as more specific
drugs, combined and sequential therapies, synthetic lethality and
immunotherapy (146). Gene editing
using clustered regularly interspaced short palindrome repeat
technology is a useful tool for identifying genes and signaling
pathways that participate in cancer drug resistance and providing
an alternative for removing resistant cells (147).
Slow progress in the field of therapy resistance is
a challenge because tumor heterogeneity, another variable that
cannot be eliminated, remains an efficient barrier underlying each
attempt to address a negative event. The tumor microenvironment is
heterogeneous with regard to the cell types, available activators
or repressors, and stromal structure and vessels. Cancer stem cells
may use different mechanisms to escape from foreign molecules or
conditions. The tumor itself contains dissimilar subpopulations
that can generate clones with new characteristics, many of which
are selectively neutral, but some have phenotypic advantages that
can result in high heterogeneity. The cells themselves may use
alternative signaling pathways for the same stimulus or vice
versa. Ultimately, therapy triggers heterogeneity, which in
turn paves the way for the development of therapy resistance,
forming a vicious circle.
However, tumor heterogeneity has an Achilles' heel.
A high mutational burden is potentially translated into
neoantigens, which are targets for immunotherapy. Although
immunotherapy has proved to be a valuable approach to treat
different cancer types, including HNSCC, it induces drug resistance
(148) and is limited to tumors
showing appropriated levels of immunogenicity. Nanomedicines
promise to circumvent this limitation, improving, for example, the
release and presentation of tumor antigens in cases with low
immunogenicity (149,150). Tissue editing approaches, which
reprogram cancer hallmarks into biologic hallmarks, may also bring
new tools to circumvent tumor heterogeneity and therapy resistance
by using continuous low-dose chemotherapy for inducing stress
responses, and transcriptional modulators for inflammation control
(151).
Conclusions
Cancer is often considered a genetic disease. In
the human body, genetic material is constantly changing and
characteristics acquired through this process may lead to genomic
instability and metabolic reprogramming of growth and survival.
These changes may not be suppressed by healthy cells.
At present, therapeutic options available for
heterogeneous tumor entities, such as HNSCC, are still limited due
to a lack of understanding of the multitude of pathways involved.
The development of novel methods to circumvent resistance and
inhibit disease progression is required, aiming to kill as many
cancer cells as possible, removing sensitive and retaining
resistant cells (152).
Further investigations are required to classify the
molecular landscape of HNSCC, and to evaluate relevant therapeutic
resistance and prognostic markers. In addition, novel therapeutic
options are required to obtain higher rates of patient survival and
reduce the levels of associated toxicity and adverse events that
may affect patient quality of life. Drug resistance studies provide
valuable insight into the mechanisms underlying cell fate pathways
and the neoplastic microenvironment. Research that utilizes
evolutionary methodological approaches may further categorize
metabolic contexts, changes in protein expression and function over
the course of the disease, diagnosis and treatment.
In conclusion, recent technological advances have
contributed to the understanding of the human genome and tumor
evolution, and have led to the identification of numerous
diagnostic and prognostic markers. However, many challenges remain
within clinical practice, including high levels of tumor
heterogeneity, undetectable clones in current assays,
drug-resistant mutations that may be pre-existing or subsequent to
therapy, identification of collateral vulnerabilities, a lack of
distinction between tumor mutations and mutations in healthy aging
cells, a lack of early disease detection and prevention, and a
limited understanding of key mechanisms of drug resistance. Novel
technologies and computational strategies coupled with further
research and clinical trials may lead to improvements in the
efficacy of cancer treatment (153–155).
Specifically regarding CTX resistance in HNSCC, the
combined use of drugs partially overcomes tumor evolution
inhibiting molecular signaling pathways crucial for the maintenance
of cancer hallmarks. In fact, current data have identified several
genomic expression profiles that correlate to resistance in HNSCC.
However, successful laboratory-based studies do not translate into
clinical applications. Novel drug delivery methods, reduced
toxicity, tailored therapies and more randomized controlled trials
are needed to develop more specific and efficient clinical
approaches to improve survival and quality of life for patients
with HNSCC.
Acknowledgements
The authors are grateful for the artwork created in
Adobe Illustrator and Adobe Photoshop by Mr. Mauro Golin, an
independent Graphic Designer based in São Paulo, SP, Brazil.
Certain data presented here were in part based upon data generated
by the TCGA Research Network: https://www.cancer.gov/tcga.
Funding
This review was supported by Fundação de Amparo à Pesquisa do
Estado de São Paulo (FAPESP) and Conselho Nacional de Pesquisas
(CNPq) grant (grant no. FAPESP 18/15468-1/EHT) and fellowships
(grant nos. FAPESP 2021/03647-1/CHDP, FAPESP 2019/07293-0/TBC,
FAPESP 2021/07087-0/TBC, CNPq 140107/2015-0/ACBS and CNPq
307327/2018-3/EHT).
Availability of data and materials
Not applicable.
Authors' contributions
CHPD contributed to the study's conception and
manuscript writing; TH, ACBS and TBC contributed to the study's
conception. EHT contributed to study conception and
manuscript-writing and review. All authors have read and approved
the final version of the manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Leemans CR, Snijders PJF and Brakenhoff
RH: The molecular landscape of head and neck cancer. Nat Rev
Cancer. 18:269–282. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cancer Genome Atlas Research Network, .
Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA,
Ellrott K, Shmulevich I, Sander C and Stuart JM: The cancer genome
atlas pan-cancer analysis project. Nat Genet. 45:1113–1120. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Leemans CR, Braakhuis BJ and Brakenhoff
RH: The molecular biology of head and neck cancer. Nat Rev Cancer.
11:9–22. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Puram SV, Mints M, Pal A, Qi Z, Reeb A,
Gelev K, Barrett TF, Gerndt S, Liu P, Parikh AS, et al: Cellular
states are coupled to genomic and viral heterogeneity in
HPV-related oropharyngeal carcinoma. Nat Genet. 55:640–650. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Johnson DE, Burtness B, Leemans CR, Lui
VWY, Bauman JE and Grandis JR: Head and neck squamous cell
carcinoma. Nat Rev Dis Primers. 6:922020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fouad S, Hauton D and D'Angiolella V:
E2F1: Cause and consequence of DNA replication stress. Front Mol
Biosci. 7:5993322021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lin M, Ji X, Lv Y, Cui D and Xie J: The
roles of TRAF3 in immune responses. Dis Markers. 2023:77878032023.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hornick EL and Bishop GA: TRAF3: Guardian
of T lymphocyte functions. Front Immunol. 14:11292512023.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Seiwert TY, Zuo Z, Keck MK, Khattri A,
Pedamallu CS, Stricker T, Brown C, Pugh TJ, Stojanov P, Cho J, et
al: Integrative and comparative genomic analysis of HPV-positive
and HPV-negative head and neck squamous cell carcinomas. Clin
Cancer Res. 21:632–641. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Häfner N, Driesch C, Gajda M, Jansen L,
Kirchmayr R, Runnebaum IB and Dürst M: Integration of the HPV16
genome does not invariably result in high levels of viral oncogene
transcripts. Oncogene. 27:1610–1617. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Parfenov M, Pedamallu CS, Gehlenborg N,
Freeman SS, Danilova L, Bristow CA, Lee S, Hadjipanayis AG, Ivanova
EV, Wilkerson MD, et al: Characterization of HPV and host genome
interactions in primary head and neck cancers. Proc Natl Acad Sci
USA. 111:15544–15549. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hettmann A, Demcsák A, Decsi G, Bach Á,
Pálinkó D, Rovó L, Nagy K, Takács M and Minarovits J: Infectious
agents associated with head and neck carcinomas. Adv Exp Med Biol.
897:63–80. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bai X, Cui C, Yin J, Li H, Gong Q, Wei B
and Lu Y: The association between oral hygiene and head and neck
cancer: A meta-analysis. Acta Odontol Scand. 81:374–395. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kitamura N, Sento S, Yoshizawa Y, Sasabe
E, Kudo Y and Yamamoto T: Current trends and future prospects of
molecular targeted therapy in head and neck squamous cell
carcinoma. Int J Mol Sci. 22:2402020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Forman R, Deshpande H, Burtness B and
Bhatia AK: Efficacy and toxicity of weekly paclitaxel, carboplatin,
and cetuximab as induction chemotherapy or in cases of metastases
or relapse for head and neck cancer with a focus on elderly or
frail patients. Head Neck. 44:1777–1786. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dokala A and Thakur SS: Extracellular
region of epidermal growth factor receptor: A potential target for
anti-EGFR drug discovery. Oncogene. 36:2337–2344. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Eze N, Lee JW, Yang DH, Zhu F, Neumeister
V, Sandoval-Schaefer T, Mehra R, Ridge JA, Forastiere A, Chung CH
and Burtness B: PTEN loss is associated with resistance to
cetuximab in patients with head and neck squamous cell carcinoma.
Oral Oncol. 91:69–78. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Emran TB, Shahriar A, Mahmud AR, Rahman T,
Abir MH, Siddiquee MF, Ahmed H, Rahman N, Nainu F, Wahyudin E, et
al: Multidrug resistance in cancer: Understanding molecular
mechanisms, immunoprevention and therapeutic approaches. Front
Oncol. 12:8916522022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Giddings EL, Champagne DP, Wu MH, Laffin
JM, Thornton TM, Valenca-Pereira F, Culp-Hill R, Fortner KA, Romero
N, East J, et al: Mitochondrial ATP fuels ABC transporter-mediated
drug efflux in cancer chemoresistance. Nat Commun. 12:28042021.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ming H, Li B, Jiang J, Qin S, Nice EC, He
W, Lang T and Huang C: Protein degradation: Expanding the toolbox
to restrain cancer drug resistance. J Hematol Oncol. 16:62023.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang N, Ma T and Yu B: Targeting
epigenetic regulators to overcome drug resistance in cancers.
Signal Transduct Target Ther. 8:692023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Su S, Chen J, Yao H, Liu J, Yu S, Lao L,
Wang M, Luo M, Xing Y, Chen F, et al:
CD10+GPR77+ cancer-associated fibroblasts
promote cancer formation and chemoresistance by sustaining cancer
stemness. Cell. 172:841–856.e16. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ito Y, Tazaki G, Kondo Y, Takahashi G and
Sakamaki F: Therapeutic effect of nintedanib on acute exacerbation
of interstitial lung diseases. Respir Med Case Rep. 26:317–320.
2019.PubMed/NCBI
|
|
25
|
Hui L and Chen Y: Tumor microenvironment:
Sanctuary of the devil. Cancer Lett. 368:7–13. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
da Cunha BR, Domingos C, Stefanini ACB,
Henrique T, Polachini GM, Castelo-Branco P and Tajara EH: Cellular
interactions in the tumor microenvironment: The role of secretome.
J Cancer. 10:4574–4587. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nia HT, Munn LL and Jain RK: Physical
traits of cancer. Science. 370:eaaz08682020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gourmet LE and Walker-Samuel S: The role
of physics in multiomics and cancer evolution. Front Oncol.
13:10680532023. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hanahan D: Hallmarks of cancer: New
dimensions. Cancer Discov. 12:31–46. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Stratton MR, Campbell PJ and Futreal PA:
The cancer genome. Nature. 458:719–724. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vendramin R, Litchfield K and Swanton C:
Cancer evolution: Darwin and beyond. EMBO J. 40:e1083892021.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nowell PC: The clonal evolution of tumor
cell populations. Science. 194:23–28. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dexter DL, Kowalski HM, Blazar BA, Fligiel
Z, Vogel R and Heppner GH: Heterogeneity of tumor cells from a
single mouse mammary tumor. Cancer Res. 38:3174–3181.
1978.PubMed/NCBI
|
|
36
|
Greaves M and Maley CC: Clonal evolution
in cancer. Nature. 481:306–313. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Swanton C: Intratumor heterogeneity:
Evolution through space and time. Cancer Res. 72:4875–4882. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Williams MJ, Werner B, Barnes CP, Graham
TA and Sottoriva A: Identification of neutral tumor evolution
across cancer types. Nat Genet. 48:238–244. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mohan M and Jagannathan N: Oral field
cancerization: An update on current concepts. Oncol Rev.
8:2442014.PubMed/NCBI
|
|
40
|
Shen X, Song S, Li C and Zhang J:
Synonymous mutations in representative yeast genes are mostly
strongly non-neutral. Nature. 606:725–731. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kristofich J, Morgenthaler AB, Kinney WR,
Ebmeier CC, Snyder DJ, Old WM, Cooper VS and Copley SD: Synonymous
mutations make dramatic contributions to fitness when growth is
limited by a weak-link enzyme. PLoS Genet. 14:e10076152018.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li R, Dong J, Zhang H, Zhao Q, Li X, Liu
X, Ye Y, Deng S, Lin D, Zheng J and Zuo Z: Clinical and genomic
characterization of neutral tumor evolution in head and neck
squamous cell carcinoma. Genomics. 112:3448–3454. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cortés-Ciriano I, Lee JJ, Xi R, Jain D,
Jung YL, Yang L, Gordenin D, Klimczak LJ, Zhang CZ, Pellman DS, et
al: Comprehensive analysis of chromothripsis in 2,658 human cancers
using whole-genome sequencing. Nat Genet. 52:331–341. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Voronina N, Wong JKL, Hübschmann D,
Hlevnjak M, Uhrig S, Heilig CE, Horak P, Kreutzfeldt S, Mock A,
Stenzinger A, et al: The landscape of chromothripsis across adult
cancer types. Nat Commun. 11:23202020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Shen MM: Chromoplexy: A new category of
complex rearrangements in the cancer genome. Cancer Cell.
23:567–569. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Baca SC, Prandi D, Lawrence MS, Mosquera
JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi
M, et al: Punctuated evolution of prostate cancer genomes. Cell.
153:666–677. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sottoriva A, Kang H, Ma Z, Graham TA,
Salomon MP, Zhao J, Marjoram P, Siegmund K, Press MF, Shibata D and
Curtis C: A Big Bang model of human colorectal tumor growth. Nat
Genet. 47:209–216. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sottoriva A, Barnes CP and Graham TA:
Catch my drift? Making sense of genomic intra-tumour heterogeneity.
Biochim Biophys Acta Rev Cancer. 1867:95–100. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Niida A, Mimori K, Shibata T and Miyano S:
Modeling colorectal cancer evolution. J Hum Genet. 66:869–878.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Laukien FH: The evolution of evolutionary
processes in organismal and cancer evolution. Prog Biophys Mol
Biol. 165:43–48. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Fearon ER and Vogelstein B: A genetic
model for colorectal tumorigenesis. Cell. 61:759–767. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Caravagna G, Giarratano Y, Ramazzotti D,
Tomlinson I, Graham TA, Sanguinetti G and Sottoriva A: Detecting
repeated cancer evolution from multi-region tumor sequencing data.
Nat Methods. 15:707–714. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
McGranahan N and Swanton C: Clonal
heterogeneity and tumor evolution: Past, present, and the future.
Cell. 168:613–628. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Niida A, Iwasaki WM and Innan H: Neutral
theory in cancer cell population genetics. Mol Biol Evol.
35:1316–1321. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Martínez-Jiménez F, Movasati A, Brunner
SR, Nguyen L, Priestley P, Cuppen E and Van Hoeck A: Pan-cancer
whole-genome comparison of primary and metastatic solid tumours.
Nature. 618:333–341. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Nguyen B, Fong C, Luthra A, Smith SA,
DiNatale RG, Nandakumar S, Walch H, Chatila WK, Madupuri R, Kundra
R, et al: Genomic characterization of metastatic patterns from
prospective clinical sequencing of 25,000 patients. Cell.
185:563–575.e11. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhao M, Liu Y, Zheng C and Qu H: dbEMT
2.0: An updated database for epithelial-mesenchymal transition
genes with experimentally verified information and precalculated
regulation information for cancer metastasis. J Genet Genomics.
46:595–597. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Acar A, Nichol D, Fernandez-Mateos J,
Cresswell GD, Barozzi I, Hong SP, Trahearn N, Spiteri I, Stubbs M,
Burke R, et al: Exploiting evolutionary steering to induce
collateral drug sensitivity in cancer. Nat Commun. 11:19232020.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Tarabichi M, Martincorena I, Gerstung M,
Leroi AM, Markowetz F; PCAWG Evolution and Heterogeneity Working
Group and Spellman PT, ; Morris QD, Lingjærde OC, Wedge DC and Van
Loo P: Neutral tumor evolution? Nat Genet. 50:1630–1633. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Persi E, Wolf YI, Horn D, Ruppin E,
Demichelis F, Gatenby RA, Gillies RJ and Koonin EV:
Mutation-selection balance and compensatory mechanisms in tumour
evolution. Nat Rev Genet. 22:251–262. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Marine JC, Dawson SJ and Dawson MA:
Non-genetic mechanisms of therapeutic resistance in cancer. Nat Rev
Cancer. 20:743–756. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Taube JH, Herschkowitz JI, Komurov K, Zhou
AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW, et
al: Core epithelial-to-mesenchymal transition interactome
gene-expression signature is associated with claudin-low and
metaplastic breast cancer subtypes. Proc Natl Acad Sci USA.
107:15449–15454. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Graves CA, Abboodi FF, Tomar S, Wells J
and Pirisi L: The translational significance of
epithelial-mesenchymal transition in head and neck cancer. Clin
Transl Med. 3:602014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Singh A and Settleman J: EMT, cancer stem
cells and drug resistance: An emerging axis of evil in the war on
cancer. Oncogene. 29:4741–4751. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Shibue T and Weinberg RA: EMT, CSCs, and
drug resistance: The mechanistic link and clinical implications.
Nat Rev Clin Oncol. 14:611–629. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Puram SV, Tirosh I, Parikh AS, Patel AP,
Yizhak K, Gillespie S, Rodman C, Luo CL, Mroz EA, Emerick KS, et
al: Single-cell transcriptomic analysis of primary and metastatic
tumor ecosystems in head and neck cancer. Cell. 171:1611–1624.e24.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Pavón MA, Arroyo-Solera I, León X,
Téllez-Gabriel M, Virós D, Gallardo A, Céspedes MV, Casanova I,
Lopez-Pousa A, Barnadas A, et al: The combined use of EFS, GPX2,
and SPRR1A expression could distinguish favorable from poor
clinical outcome among epithelial-like head and neck carcinoma
subtypes. Head Neck. 41:1830–1845. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
DeCamp SJ, Tsuda VMK, Ferruzzi J, Koehler
SA, Giblin JT, Roblyer D, Zaman MH, Weiss ST, Kılıç A, De Marzio M,
et al: Epithelial layer unjamming shifts energy metabolism toward
glycolysis. Sci Rep. 10:183022020. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
De Marzio M, Kılıç A, Maiorino E, Mitchel
JA, Mwase C, O'Sullivan MJ, McGill M, Chase R, Fredberg JJ, Park
JA, et al: Genomic signatures of the unjamming transition in
compressed human bronchial epithelial cells. Sci Adv.
7:eabf10882021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Kılıç A, Ameli A, Park JA, Kho AT,
Tantisira K, Santolini M, Cheng F, Mitchel JA, McGill M, O'Sullivan
MJ, et al: Mechanical forces induce an asthma gene signature in
healthy airway epithelial cells. Sci Rep. 10:9662020. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Ataie-Kachoie P, Pourgholami MH,
Richardson DR and Morris DL: Gene of the month: Interleukin 6
(IL-6). J Clin Pathol. 67:932–937. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Taher MY, Davies DM and Maher J: The role
of the interleukin (IL)-6/IL-6 receptor axis in cancer. Biochem Soc
Trans. 46:1449–1462. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Wu Y and Zhou BP:
TNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and
invasion. Br J Cancer. 102:639–644. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Dongre A and Weinberg RA: New insights
into the mechanisms of epithelial-mesenchymal transition and
implications for cancer. Nat Rev Mol Cell Biol. 20:69–84. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Li Y, Azmi AS and Mohammad RM: Deregulated
transcription factors and poor clinical outcomes in cancer
patients. Semin Cancer Biol. 86:122–134. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Xin W, Zhao C, Jiang L, Pei D, Zhao L and
Zhang C: Identification of a novel epithelial-mesenchymal
transition gene signature predicting survival in patients with
HNSCC. Pathol Oncol Res. 27:5851922021. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Vallina C, López-Pintor RM,
González-Serrano J, de Vicente JC, Hernández G and Lorz C: Genes
involved in the epithelial-mesenchymal transition in oral cancer: A
systematic review. Oral Oncol. 117:1053102021. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Okuyama K, Suzuki K and Yanamoto S:
Relationship between tumor budding and partial
epithelial-mesenchymal transition in head and neck cancer. Cancers
(Basel). 15:11112023. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Tamimi A, Tamimi A, Sorkheh F, Asl SM,
Ghafari A, Karimi AG, Erabi G, Pourmontaseri H and Deravi N:
Monoclonal antibodies for the treatment of squamous cell carcinoma:
A literature review. Cancer Rep (Hoboken). 6:e18022023. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Byeon HK, Ku M and Yang J: Beyond EGFR
inhibition: Multilateral combat strategies to stop the progression
of head and neck cancer. Exp Mol Med. 51:1–14. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Klein P, Mattoon D, Lemmon MA and
Schlessinger J: A structure-based model for ligand binding and
dimerization of EGF receptors. Proc Natl Acad Sci USA. 101:929–934.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Nair S, Trummell HQ, Rajbhandari R, Thudi
NK, Nozell SE, Warram JM, Willey CD, Yang ES, Placzek WJ, Bonner JA
and Bredel M: Novel EGFR ectodomain mutations associated with
ligand-independent activation and cetuximab resistance in head and
neck cancer. PLoS One. 15:e02290772020. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Purba ER, Saita EI and Maruyama IN:
Activation of the EGF receptor by ligand binding and oncogenic
mutations: The ‘rotation model’. Cells. 6:132017. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Kriegs M, Clauditz TS, Hoffer K, Bartels
J, Buhs S, Gerull H, Zech HB, Bußmann L, Struve N, Rieckmann T, et
al: Analyzing expression and phosphorylation of the EGF receptor in
HNSCC. Sci Rep. 9:135642019. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Kalyankrishna S and Grandis JR: Epidermal
growth factor receptor biology in head and neck cancer. J Clin
Oncol. 24:2666–2672. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Najafi M, Ahmadi A and Mortezaee K:
Extracellular-signal-regulated kinase/mitogen-activated protein
kinase signaling as a target for cancer therapy: An updated review.
Cell Biol Int. 43:1206–1222. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Nishihara S, Yamaoka T, Ishikawa F,
Higuchi K, Hasebe Y, Manabe R, Kishino Y, Kusumoto S, Ando K,
Kuroda Y, et al: Mechanisms of EGFR-TKI-induced apoptosis and
strategies targeting apoptosis in EGFR-mutated non-small cell lung
cancer. Genes (Basel). 13:21832022. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Liu X, Mei W, Zhang P and Zeng C: PIK3CA
mutation as an acquired resistance driver to EGFR-TKIs in non-small
cell lung cancer: Clinical challenges and opportunities. Pharmacol
Res. 202:1071232024. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Lai SY and Johnson FM: Defining the role
of the JAK-STAT pathway in head and neck and thoracic malignancies:
Implications for future therapeutic approaches. Drug Resist Updat.
13:67–78. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Li L, Ji S, Shrestha C, Jiang Y, Liao L,
Xu F, Liu Z, Bikle DD and Xie Z: p120-catenin suppresses
proliferation and tumor growth of oral squamous cell carcinoma via
inhibiting nuclear phospholipase C-γ1 signaling. J Cell Physiol.
235:9399–9413. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Mittal S, Kamath A, Joseph AM and Rajala
MS: PLCγ1-dependent invasion and migration of cells expressing
NSCLC-associated EGFR mutants. Int J Oncol. 57:989–1000.
2020.PubMed/NCBI
|
|
94
|
Li Q, Tie Y, Alu A, Ma X and Shi H:
Targeted therapy for head and neck cancer: Signaling pathways and
clinical studies. Signal Transduct Target Ther. 8:312023.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Silva-Oliveira RJ, Melendez M, Martinho O,
Zanon MF, de Souza Viana L, Carvalho AL and Reis RM: AKT can
modulate the in vitro response of HNSCC cells to irreversible EGFR
inhibitors. Oncotarget. 8:53288–53301. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Kordbacheh F and Farah CS: Current and
emerging molecular therapies for head and neck squamous cell
carcinoma. Cancers (Basel). 13:54712021. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Ebrahimi N, Fardi E, Ghaderi H, Palizdar
S, Khorram R, Vafadar R, Ghanaatian M, Rezaei-Tazangi F, Baziyar P,
Ahmadi A, et al: Receptor tyrosine kinase inhibitors in cancer.
Cell Mol Life Sci. 80:1042023. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Lacas B, Carmel A, Landais C, Wong SJ,
Licitra L, Tobias JS, Burtness B, Ghi MG, Cohen EEW, Grau C, et al:
Meta-analysis of chemotherapy in head and neck cancer (MACH-NC): An
update on 107 randomized trials and 19,805 patients, on behalf of
MACH-NC group. Radiother Oncol. 156:281–293. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Vermorken JB, Mesia R, Rivera F, Remenar
E, Kawecki A, Rottey S, Erfan J, Zabolotnyy D, Kienzer HR, Cupissol
D, et al: Platinum-based chemotherapy plus cetuximab in head and
neck cancer. N Engl J Med. 359:1116–1127. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Guigay J, Aupérin A, Fayette J,
Saada-Bouzid E, Lafond C, Taberna M, Geoffrois L, Martin L,
Capitain O, Cupissol D, et al: Cetuximab, docetaxel, and cisplatin
versus platinum, fluorouracil, and cetuximab as first-line
treatment in patients with recurrent or metastatic head and neck
squamous-cell carcinoma (GORTEC 2014-01 TPExtreme): A multicentre,
open-label, randomised, phase 2 trial. Lancet Oncol. 22:463–475.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Eggers H, Häbel L, Ganser A, Grünwald V,
Merten R, Warnecke A, Durisin M and Ivanyi P: Anti-EGFR-based
therapy in recurrent or metastatic HNSCC-what difference does it
make? Cancer Invest. 41:93–100. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Rich TA, Shepard RC and Mosley ST: Four
decades of continuing innovation with fluorouracil: Current and
future approaches to fluorouracil chemoradiation therapy. J Clin
Oncol. 22:2214–2232. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Muraro E, Fanetti G, Lupato V, Giacomarra
V, Steffan A, Gobitti C, Vaccher E and Franchin G: Cetuximab in
locally advanced head and neck squamous cell carcinoma: Biological
mechanisms involved in efficacy, toxicity and resistance. Crit Rev
Oncol Hematol. 164:1034242021. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Okada Y, Kimura T, Nakagawa T, Okamoto K,
Fukuya A, Goji T, Fujimoto S, Sogabe M, Miyamoto H, Muguruma N, et
al: EGFR downregulation after Anti-EGFR therapy predicts the
antitumor effect in colorectal cancer. Mol Cancer Res.
15:1445–1454. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Kang JJ, Ko A, Kil SH, Mallen-St Clair J,
Shin DS, Wang MB and Srivatsan ES: EGFR pathway targeting drugs in
head and neck cancer in the era of immunotherapy. Biochim Biophys
Acta Rev Cancer. 1878:1888272023. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Chaudhary R, Slebos RJC, Noel LC, Song F,
Poole MI, Hoening DS, Hernandez-Prera JC, Conejo-Garcia JR,
Guevara-Patino JA, Wang X, et al: EGFR inhibition by cetuximab
modulates hypoxia and IFN response genes in head and neck squamous
cell carcinoma. Cancer Res Commun. 3:896–907. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Bugaj LJ, Sabnis AJ, Mitchell A, Garbarino
JE, Toettcher JE, Bivona TG and Lim WA: Cancer mutations and
targeted drugs can disrupt dynamic signal encoding by the Ras-Erk
pathway. Science. 361:eaao30482018. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Jie HB, Schuler PJ, Lee SC, Srivastava RM,
Argiris A, Ferrone S, Whiteside TL and Ferris RL: CTLA-4+
regulatory T cells increased in cetuximab-treated head and neck
cancer patients suppress NK cell cytotoxicity and correlate with
poor prognosis. Cancer Res. 75:2200–2210. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Kagohara LT, Zamuner F, Davis-Marcisak EF,
Sharma G, Considine M, Allen J, Yegnasubramanian S, Gaykalova DA
and Fertig EJ: Integrated single-cell and bulk gene expression and
ATAC-seq reveals heterogeneity and early changes in pathways
associated with resistance to cetuximab in HNSCC-sensitive cell
lines. Br J Cancer. 123:101–113. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Ryman JT and Meibohm B: Pharmacokinetics
of monoclonal antibodies. CPT Pharmacometrics Syst Pharmacol.
6:576–588. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Tathineni P, Joshi N and Jelinek MJ:
Current state and future directions of EGFR-directed therapy in
head and neck cancer. Curr Treat Options Oncol. 24:680–692. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
de Castro G Jr, Alves GV, Castro AF,
Chaves ALF, De Marchi P, de Oliveira TB, Dias FL, Guindalini RSC,
Nicolau UR, Soares A and Mora PAR: Criteria for eligibility to
cisplatin in the curative treatment of head and neck cancer:
Consensus opinion from a panel of experts. Crit Rev Oncol Hematol.
131:30–34. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Carinato H, Burgy M, Ferry R, Fischbach C,
Kalish M, Guihard S, Brahimi Y, Flesch H, Bronner G, Schultz P, et
al: Weekly paclitaxel, carboplatin, and cetuximab as first-line
treatment of recurrent and/or metastatic head and neck squamous
cell carcinoma for patients ineligible to cisplatin-based
chemotherapy: A retrospective monocentric study in 60 patients.
Front Oncol. 11:7145512021. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Abdulla M, Belal AA, Sakr A, El Arab LE,
Mokhtar M, Allahloubi N, Ghali R, Hashem T and Arafat W:
Eligibility criteria to cisplatin in head and neck squamous cell
carcinoma: Egyptian expert opinion. Health Sci Rep. 6:e10372023.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Guigay J, Fayette J, Dillies AF, Sire C,
Kerger JN, Tennevet I, Machiels JP, Zanetta S, Pointreau Y, Bozec
Le Moal L, et al: Cetuximab, docetaxel, and cisplatin as first-line
treatment in patients with recurrent or metastatic head and neck
squamous cell carcinoma: A multicenter, phase II GORTEC study. Ann
Oncol. 26:1941–1947. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Mehanna H, Robinson M, Hartley A, Kong A,
Foran B, Fulton-Lieuw T, Dalby M, Mistry P, Sen M, O'Toole L, et
al: Radiotherapy plus cisplatin or cetuximab in low-risk human
papillomavirus-positive oropharyngeal cancer (De-ESCALaTE HPV): An
open-label randomised controlled phase 3 trial. Lancet. 393:51–60.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Silver JA, Turkdogan S, Roy CF,
Subramaniam T, Henry M and Sadeghi N: De-escalation strategies for
human papillomavirus-associated oropharyngeal squamous cell
carcinoma-where are we now? Curr Oncol. 29:3668–3697. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Willey CD, Anderson JC, Trummell HQ, Naji
F, de Wijn R, Yang ES, Bredel M, Thudi NK and Bonner JA:
Differential escape mechanisms in cetuximab-resistant head and neck
cancer cells. Biochem Biophys Res Commun. 517:36–42. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Yonesaka K, Tanaka K, Kitano M, Kawakami
H, Hayashi H, Takeda M, Sakai K, Nishio K, Doi K and Nakagawa K:
Aberrant HER3 ligand heregulin-expressing head and neck squamous
cell carcinoma is resistant to anti-EGFR antibody cetuximab, but
not second-generation EGFR-TKI. Oncogenesis. 8:542019. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Picon H and Guddati AK: Mechanisms of
resistance in head and neck cancer. Am J Cancer Res. 10:2742–2751.
2020.PubMed/NCBI
|
|
122
|
Ortiz-Cuaran S, Bouaoud J, Karabajakian A,
Fayette J and Saintigny P: Precision medicine approaches to
overcome resistance to therapy in head and neck cancers. Front
Oncol. 11:6143322021. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Cheng H, Fertig EJ, Ozawa H, Hatakeyama H,
Howard JD, Perez J, Considine M, Thakar M, Ranaweera R, Krigsfeld G
and Chung CH: Decreased SMAD4 expression is associated with
induction of epithelial-to-mesenchymal transition and cetuximab
resistance in head and neck squamous cell carcinoma. Cancer Biol
Ther. 16:1252–1258. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Boeckx C, Blockx L, Op de Beeck K, Limame
R, Camp GV, Peeters M, Vermorken JB, Specenier P, Wouters A, Baay M
and Lardon F: Establishment and characterization of cetuximab
resistant head and neck squamous cell carcinoma cell lines: Focus
on the contribution of the AP-1 transcription factor. Am J Cancer
Res. 5:1921–1938. 2015.PubMed/NCBI
|
|
125
|
Citron F, Segatto I, Musco L, Pellarin I,
Rampioni Vinciguerra GL, Franchin G, Fanetti G, Miccichè F,
Giacomarra V, Lupato V, et al: miR-9 modulates and predicts the
response to radiotherapy and EGFR inhibition in HNSCC. EMBO Mol
Med. 13:e128722021. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Morvan VL, Richard É, Cadars M, Fessart D,
Broca-Brisson L, Auzanneau C, Pasquies A, Modesto A, Lusque A,
Mathoulin-Pélissier S, et al: Cytochrome P450 1B1 polymorphism
drives cancer cell stemness and patient outcome in head-and-neck
carcinoma. Br J Cancer. 123:772–784. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Wheeler DL, Huang S, Kruser TJ,
Nechrebecki MM, Armstrong EA, Benavente S, Gondi V, Hsu KT and
Harari PM: Mechanisms of acquired resistance to cetuximab: Role of
HER (ErbB) family members. Oncogene. 27:3944–3956. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Nelhűbel GA, Cserepes M, Szabó B, Türk D,
Kárpáti A, Kenessey I, Rásó E, Barbai T, Hegedűs Z, László V, et
al: EGFR alterations influence the cetuximab treatment response and
c-MET tyrosine-kinase inhibitor sensitivity in experimental head
and neck squamous cell carcinomas. Pathol Oncol Res. 27:6202562021.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Zhang Y, Xia M, Jin K, Wang S, Wei H, Fan
C, Wu Y, Li X, Li X, Li G, et al: Function of the c-Met receptor
tyrosine kinase in carcinogenesis and associated therapeutic
opportunities. Mol Cancer. 17:452018. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Cortesina G, Martone T, Galeazzi E,
Olivero M, De Stefani A, Bussi M, Valente G, Comoglio PM and Di
Renzo MF: Staging of head and neck squamous cell carcinoma using
the MET oncogene product as marker of tumor cells in lymph node
metastases. Int J Cancer. 89:286–292. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Slomiany MG, Black LA, Kibbey MM, Tingler
MA, Day TA and Rosenzweig SA: Insulin-like growth factor-1 receptor
and ligand targeting in head and neck squamous cell carcinoma.
Cancer Lett. 248:269–279. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Gonzales CB, De La Chapa JJ, Saikumar P,
Singha PK, Dybdal-Hargreaves NF, Chavez J, Horning AM, Parra J and
Kirma NB: Co-targeting ALK and EGFR parallel signaling in oral
squamous cell carcinoma. Oral Oncol. 59:12–19. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Iyer G, Price J, Bourgeois S, Armstrong E,
Huang S and Harari PM: Insulin-like growth factor 1 receptor
mediated tyrosine 845 phosphorylation of epidermal growth factor
receptor in the presence of monoclonal antibody cetuximab. BMC
Cancer. 16:7732016. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Krumbach R, Schüler J, Hofmann M,
Giesemann T, Fiebig HH and Beckers T: Primary resistance to
cetuximab in a panel of patient-derived tumour xenograft models:
Activation of MET as one mechanism for drug resistance. Eur J
Cancer. 47:1231–1243. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Ouyang X, Barling A, Lesch A, Tyner JW,
Choonoo G, Zheng C, Jeng S, West TM, Clayburgh D, Courtneidge SA,
et al: Induction of anaplastic lymphoma kinase (ALK) as a novel
mechanism of EGFR inhibitor resistance in head and neck squamous
cell carcinoma patient-derived models. Cancer Biol Ther.
19:921–933. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Umemori K, Ono K, Eguchi T, Kawai H,
Nakamura T, Ogawa T, Yoshida K, Kanemoto H, Sato K, Obata K, et al:
EpEX, the soluble extracellular domain of EpCAM, resists cetuximab
treatment of EGFR-high head and neck squamous cell carcinoma. Oral
Oncol. 142:1064332023. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Gires O, Pan M, Schinke H, Canis M and
Baeuerle PA: Expression and function of epithelial cell adhesion
molecule EpCAM: Where are we after 40 years? Cancer Metastasis Rev.
39:969–987. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Went PT, Lugli A, Meier S, Bundi M,
Mirlacher M, Sauter G and Dirnhofer S: Frequent EpCam protein
expression in human carcinomas. Hum Pathol. 35:122–128. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Mignion L, Acciardo S, Gourgue F, Joudiou
N, Caignet X, Goebbels RM, Corbet C, Feron O, Bouzin C, Cani PD, et
al: Metabolic imaging using hyperpolarized pyruvate-lactate
exchange assesses response or resistance to the EGFR inhibitor
cetuximab in patient-derived HNSCC xenografts. Clin Cancer Res.
26:1932–1943. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Bae T, Hallis SP and Kwak MK: Hypoxia,
oxidative stress, and the interplay of HIFs and NRF2 signaling in
cancer. Exp Mol Med. 56:501–514. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Deng L, Wang L, Zhang J, Zhao L, Meng Y,
Zheng J, Xu W, Zhu Z and Huang H: The mechanism of action and
biodistribution of a novel EGFR/VEGF bispecific fusion protein that
exhibited superior antitumor activities. Heliyon. 9:e169222023.
View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Liang W, Zheng Y, Zhang J and Sun X:
Multiscale modeling reveals angiogenesis-induced drug resistance in
brain tumors and predicts a synergistic drug combination targeting
EGFR and VEGFR pathways. BMC Bioinformatics. 20 (Suppl 7):S2032019.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Wiechec E, Hansson KT, Alexandersson L,
Jönsson JI and Roberg K: Hypoxia mediates differential response to
anti-EGFR therapy in HNSCC cells. Int J Mol Sci. 18:9432017.
View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Ge H, Ferris RL and Wang JH: Cetuximab
responses in patients with HNSCC correlate to clonal expansion
feature of peripheral and tumor-infiltrating T cells with Top
T-cell receptor clonotypes. Clin Cancer Res. 29:647–658. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Parikh AS, Yu VX, Flashner S, Okolo OB, Lu
C, Henick BS, Momen-Heravi F, Puram SV, Teknos T, Pan Q and
Nakagawa H: Patient-derived three-dimensional culture techniques
model tumor heterogeneity in head and neck cancer. Oral Oncol.
138:1063302023. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Cree IA and Charlton P: Molecular chess?
Hallmarks of anti-cancer drug resistance. BMC Cancer. 17:102017.
View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Shirani-Bidabadi S, Tabatabaee A, Tavazohi
N, Hariri A, Aref AR, Zarrabi A, Casarcia N, Bishayee A and Mirian
M: CRISPR technology: A versatile tool to model, screen, and
reverse drug resistance in cancer. Eur J Cell Biol. 102:1512992023.
View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Liu S, Wang R and Fang J: Exploring the
frontiers: Tumor immune microenvironment and immunotherapy in head
and neck squamous cell carcinoma. Discov Oncol. 15:222024.
View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Jin Y, Huang Y, Ren H, Huang H, Lai C,
Wang W, Tong Z, Zhang H, Wu W, Liu C, et al: Nano-enhanced
immunotherapy: Targeting the immunosuppressive tumor
microenvironment. Biomaterials. 305:1224632024. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Avgoustakis K and Angelopoulou A:
Biomaterial-based responsive nanomedicines for targeting solid
tumor microenvironments. Pharmaceutics. 16:1792024. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Harrer DC, Lüke F, Pukrop T, Ghibelli L,
Reichle A and Heudobler D: Addressing genetic tumor heterogeneity,
post-therapy metastatic spread, cancer repopulation, and
development of acquired tumor cell resistance. Cancers (Basel).
16:1802023. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Derbal Y: Cell adaptive fitness and cancer
evolutionary dynamics. Cancer Inform. 22:117693512311546792023.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Parseghian CM, Napolitano S, Loree JM and
Kopetz S: Mechanisms of innate and acquired resistance to anti-EGFR
therapy: A review of current knowledge with a focus on rechallenge
therapies. Clin Cancer Res. 25:6899–6908. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Walens A, Lin J, Damrauer JS, McKinney B,
Lupo R, Newcomb R, Fox DB, Mabe NW, Gresham J, Sheng Z, et al:
Adaptation and selection shape clonal evolution of tumors during
residual disease and recurrence. Nat Commun. 11:50172020.
View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Fittall MW and Van Loo P: Translating
insights into tumor evolution to clinical practice: Promises and
challenges. Genome Med. 11:202019. View Article : Google Scholar : PubMed/NCBI
|