Introduction
Asthma is one of the most common chronic diseases
characterized by airway hyperresponsiveness (AHR) and airway
remodeling. It has been well established that airway smooth muscle
(ASM) cells are the main components of the respiratory tract. They
are believed to have a role in the pathogenesis of asthma through
their contractile properties (1).
Additionally, it is widely accepted that the cells act as
immunomodulation, which contribute to the inflammation of the
respiratory airway and structural alterations via inflammatory and
immunological factors associated with asthma (2). Noble et al (3) proposed that ASM contraction, in
combination with cellular mechanotransduction and novel
contraction-inflammation synergies, contributed to the
heterogeneous pathogenesis of asthma. The contraction is the basis
of ASM function. It is well known that ASM contraction is regulated
by secondary messengers, such as guanosine 3′,5′-cyclic phosphate,
cyclic adenosine monophosphate and Ca2+ (4). Among them, Ca2+ is an
important secondary messenger that regulates miscellaneous
responses in ASM cells, such as contraction, relaxation,
proliferation, migration and cytokine secretion. Elevation of the
Ca2+ level is derived from intracellular Ca2+
release out of the sarcoplasmic reticulum (SR) and extracellular
Ca2+ influx (5,6). Wang et al (7) identified that the change of cytosolic
Ca2+ level determined the primary-signal-regulating
contractile function of ASM cells. It is clear that Ca2+
is a key factor for assessing the efficacy of drugs used in
asthma.
Mabuterol hydrochloride (Mab) (Fig. 1) as a novel β2-agonist with
high selectivity has good pharmacokinetic properties, such as an
orally complete absorption and a long duration of action, and it
has been clinically used as a bronchodilator in the treatment of
asthma (8). Pharmacodynamic studies of
Mab have been conducted since it was first synthesized by German
scholars in 1984. Osada et al (9) studied the effect of Mab on the
cardiovascular system and smooth muscle organs of rats, cats and
dogs and made a comparison with those of isoprenaline, salbutamol
and procaterol. They found that the drug did not influence
α-adrenergic, acetylcholine (Ach) and histamine receptors, and was
a specific β2 blocker with no β1-stimulation.
The effect on blood pressure and peripheral vascular resistance in
dogs was 365 and 118 times less compared to isoprenaline.
Additionally, it was shown in the study by Akahane et al
(10) that Mab, when injected into the
sinus node artery of the isolated atrium, dose-dependently
increased the atrial rate and contractile force, which were
inhibited by a selective β2-receptor antagonist, ICI
118551, and only slightly attenuated by atenolol. These
weak-positive chronotropic and inotropic effects were clearly
produced by stimulating β2-adrenoceptors on the perfused
canine right atrium. However, there is limited literature regarding
the precise mechanism of action for Mab.
In the present study, a renewed and stable method of
culturing guinea pig ASM cells was established. The suppression of
increasing intracellular calcium by Mab was investigated with
several detection methods and two agents Fura-2/AM, as well as
Fluo-3/AM as a Ca2+ indicator.
Materials and methods
Animals
Male or female Hartley guinea pigs, weighing 150–200
g, were provided by the Experimental Animal Center of Shenyang
Pharmaceutical University (Shenyang, Liaoning, China). Animals were
bred in a facility controlled by temperature (26±3°C), relative
humidity (50±5%) and light (14 and 10 h of light and dark), with
free access to food and water, with added vitamin C. All the
experimental procedures in the present study were carried out in
accordance with the Internationally Accepted Principles and the
Guidelines for the Care and Use of Animal Center of Shenyang
Pharmaceutical University.
Drugs and chemicals
Mab was supplied by the Pharmaceutical Engineering
Department, Shenyang Pharmaceutical University (enantiomeric excess
>99%). Ach was purchased from Sinopharm Chemical Reagent Co.,
Ltd. (Shanghai, China). Dulbecco's modified Eagle's medium (DMEM)
and Hanks' balanced salt solution (HBSS) were purchased from
Gibco-BRL (Carlsbad, CA, USA) and type I collagenase from Beijing
Solarbio Science and Technology Co., Ltd. (Beijing, China). Fetal
bovine serum (FBS) was produced by Tianjin Hualida Biotechnology
Co., Ltd. (Tianjin, China). Triton X-100 and
3-(4,5-dimethylthinazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
were obtained from Amresco LLC (Solon, OH, USA). Mouse
anti-α-smooth muscle actin (α-SMA), 5% bovine serum albumin (BSA),
streptavidin-biotin complex (SABC) immunohistochemical staining kit
and 3,3′ diaminobenzidine (DAB) chromogenic reagent kit were all
purchased from Wuhan Boster Biological Technology Co., Ltd. (Wuhan,
China). Fura-2/AM, Fluo-3/AM and fluorescein isothiocyanate
(FITC)-labeled goat anti-mouse immunoglobulin G (IgG) were from
Beyotime Institute of Biotechnology (Haimen, Jiangsu, China).
Applying two different methods to the
culture
One of the methods used collagenase to pretreat
tracheal tissues (CPTT) and the other did not. Freshly dispersed
tracheal smooth muscle strips of guinea pig were prepared as
described previously (11). Briefly,
tracheal samples of several guinea pigs were mechanically isolated
and instantly placed in 4°C HBSS. They were dissected free of
adhering fat and other connective tissues. Smooth muscle strips
without any tracheal cartilage were obtained and cut into
1-mm3 pieces. The ASM pieces were randomly divided into
two groups. Half of the pieces were moved into one culture flask
with a cell growth area of 25 cm2 and put evenly on the
inside wall. The culture process followed the steps of the
traditional method. The other half were placed into another flask
after digestion with 2% type I collagenase for 20 min (37°C, 5%
CO2) and the culture process followed the steps of the
CPTT method. A total of 2 ml of DMEM containing 20% FBS, 100 IU/ml
penicillin, 100 IU/ml streptomycin and 2 mmol/l L-glutamine was
added to immerse the ASM pieces when the edge of ASM had dried and
began to tightly attach to the surface of the flask. All the flasks
were placed in the humidified atmosphere containing 5%
CO2 at 37°C and the medium was partially replaced every
3 days in accordance with the routine procedure in cell culture.
Cell growth was observed daily. The time for the cells to initially
migrate out of the ASM pieces as well as the culture having to be
generated due to the thick density of the cells in the flask was
recorded. The pieces, out of which ASM cells first migrate, were
moved into a new flask when enough cells were harvested. They could
be repetitively used in the production of the ASM cells ≤3
times.
Cell viability assay
When the cell density was ~80%, the ASM pieces were
moved into another culture flask to be fully prepared as described
above. Subsequently, the cells were detached with the mixed
solution of 0.25% trypsin and 0.02% EDTA at 37°C for 3 min in
preparation for cell generation. The MTT assay was used to
determine cell viability (12).
Briefly, 500 µl of the third generation of cell suspension at a
density of 1×104 cells/ml was seeded in 96-well plates
and incubated at 37°C with 5% CO2 for 1, 2, 3, 4, 5, 6
and 7 days, respectively. When the MTT assay was conducted, 150 µl
of phosphate-buffered saline (PBS) with 0.5 mg/ml MTT was added to
the medium in each well and incubated at 37°C with 5%
CO2 for 4 h. Subsequently, the supernatant was removed
by aspiration and dimethyl sulfoxide was added into each well. The
reaction was sustained for 10 min at room temperature. The amount
of MTT formazan was quantified by measuring optical density (OD) at
492 nm with a Varioskan Flash (Thermo Fisher Scientific, Inc.,
Rockford, IL, USA). A cell growth curve was generated with GraphPad
Prism 5 (GraphPad Software, San Diego, CA, USA).
Identification of guinea pig ASM
cells
To confirm that the cells were ASM cells and not
epithelial cells or fibroblasts, homogeneity was confirmed with
α-SMA according to a previously described method (13). Briefly, ASM cells of generation three,
four or five were cultivated in a 24-well plate at a density of
10,000 cells/well. They were rinsed with 0.01 M PBS (pH 7.2–7.4),
fixed with 4% phosphate-buffered paraformaldehyde for 30 min, and
attached to the bottom of the plate and grew in a good condition.
Subsequently, 5% BSA was added to block the non-specific proteins
for 20 min after they were treated with 0.25% Triton X-100 for 10
min at room temperature. The cells were incubated with α-SMA
(1:200) in a wet box at 4°C overnight. Following this, they were
rinsed 3 times with 0.01 M PBS. The plate used for
immunocytochemistry was incubated with goat anti-mouse IgG (1:200)
for 20 min, treated with SABC for 30 min at 37°C and the
chromogenic reaction was conducted with DAB for 8 min. The image
was observed under an inverted system microscope (IX71; Olympus,
Tokyo, Japan). The plate used for immunofluorescence was incubated
with FITC-labeled goat anti-mouse IgG for 60 min at 37°C. The
immunofluorescent image was observed under the inverted microscope
with an absorption peak at 492 nm and emission peak at 520 nm, and
the data were saved.
Determination of intracellular
Ca2+
Measurement with Fura-2/AM
Intracellular Ca2+ was indicated with a
fluorescent molecular probe, Fura-2/AM, as described previously
(14). In brief, the cells were
carefully moved into a sterile eppendorf tube at a density of
~2×105 cells/tube. Subsequently, they were preloaded
with Fura-2/AM for 60 min in a humidified incubator (37°C, 5%
CO2) at a final concentration of 5 µmol/l in DMEM (pH
7.2–7.4) supplemented with 10% FBS. The cells loaded with Fura-2/AM
were rinsed with 0.2% BSA twice. The eppendorf tube was centrifuged
at 220 × g for 5 min at room temperature and HBSS without
Ca2+ was used to suspend the cell pellets. One section
of the cells was loaded with Fura-2/AM in 100 µl of suspension and
was moved into black 96-well culture plates (Corning Life Sciences,
Tewksbury MA, USA) at the density of ~2×104 cells/well
for the determination of Ca2+ fluorescence intensity (F)
with the Varioskan Flash under the condition of an excitation
wavelength at 340 nm and emission wavelength at 510 nm. The
inhibitory rate of calcium was calculated according to the
equation: Calcium inhibitory rate (%) = (F340 control −
F340 mabuterol)/F340control × 100%. A second
section of the cells was loaded with Fura-2/AM in 200 µl of
suspension and was transferred into 24-well culture plates to
obtain Ca2+ fluorescent images at the ultraviolet region
under an inverted system microscope (IX71; Olympus).
Measurement with Fluo-3/AM
A total of 2 µmol/l of the Ca2+-sensitive
Fluo-3/AM was required according to the manufacturer's instruction
when the intracellular Ca2+ level was determined with
flow cytometry. Cells at the density of 3×106/ml were
incubated at 37°C for 45 min in the dark after the treatment with
Mab plus Ach, as described previously. The cells were gently rinsed
with HBSS without Ca2+ 3 times. When Fluo-3/AM binds to
cytoplasmic-free calcium, the complex emits green fluorescence
under the stimulation of the 488 nm line of an argon ion laser. The
fluorescent intensity at 525 nm was determined at 37°C with the
Becton-Dickinson Immunocytometry system (FACSCalibur; BD
Biosciences, San Jose, CA, USA) and the light signal was converted
into an electric signal with linear amplification.
Statistical analysis
Results are expressed as mean ± standard error of
the mean and statistical comparisons among groups were performed
with one-way analysis of variance followed by least significant
difference or independent samples t-test using SPSS 16.0 (SPSS
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference in all the experiments. Figure
plotting was conducted with the aid of software GraphPad Prism 5
(GraphPad Software Inc., San Diego, CA, USA).
Results
CPTT method for an efficient culture
of ASM cells
As is shown in Fig. 2,
the number of days for the ASM cells to start migrating out of the
‘tissue blocks’ (Fig. 2A) were
significantly different between the groups treated with the two
methods. The number of days for the culture to generate, as it
comprised too many cells (Fig. 2B),
appeared to be different between the two groups. The average time
was 4.2 days for the cells to start growing out in the culture
pretreated with collagenase, which was less than that (6.4 days) in
the culture without pretreatment with the enzyme. The time for the
dense cells to be initially passed was 5.4 days in the collagenase
group and 6.8 days in the collagenase-free group, which also showed
a high efficacy with the CPTT method. The status of the cells in
the two groups on day 6 after the guinea pig tracheal smooth muscle
strip was planted is shown in Fig. 2C and
D. The ASM cells in Fig. 2C were
evidently thick, with a density ~80%, while those in Fig. 2D were just beginning to migrate out of
the pieces at the same time. In addition, the status of the ASM
cells migrating from the second-hand pieces treated with the CPTT
method was better than that of the traditional method (Fig. 2E and F).
Morphology and viability of ASM
cells
Cells migrating from the ASM pieces treated with
collagenase began to attach to the surface of the culture flask 6 h
after they were generated and spread out gradually in the following
2 days. Their morphology was expressed in fusiform shown with
arrowheads or an irregular triangle shown with arrows in Fig. 3A. The density of the cells became
significantly thick on day 4 and they were in a good state
(Fig. 3B). The typical peak-valley
pattern of ASM cells was observed under an optical microscope ~day
6. However, this was at the same time that the cells aligned so
closely that their morphology looked abnormal in certain local
areas, which is shown with arrows in Fig.
3C.
Cell viability was determined with the MTT assay at
days 1 to 7, respectively, after they were generated. As was
observed in the growth curve of Fig.
3D, OD values increased significantly on days 4 and 6
(P<0.05), which indicated that the cells proliferated
significantly.
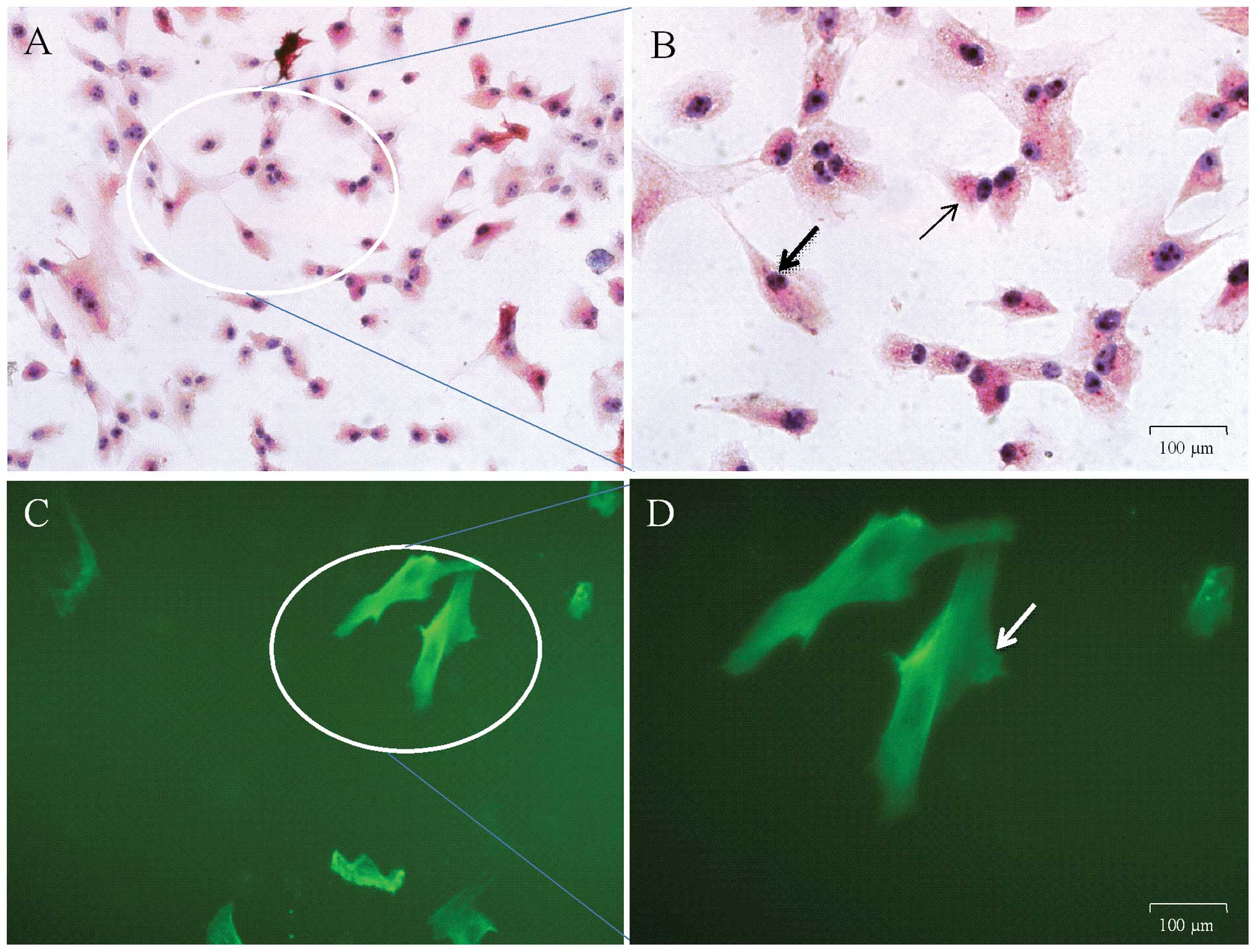
Identification of ASM cells
Guinea pig ASM cells were identified with
immunocytochemistry and immunofluorescent staining subsequently to
being loaded with the specific α-SMA antibody. The results of
immunocytochemistry were as stated in Fig.
4A and B. The magnified cells in Fig.
4 were in various shapes, including the irregular triangle form
indicated with thin arrows and fusiform with common arrows. Green
fluorescence could be observed at 492 nm under an inverted
fluorescent microscope once the cells were loaded with FITC for
identification with immunofluorescent staining, as expressed in
Fig. 4C and D. The arrow in Fig. 4D indicates ASM cells in the fusiform.
It was found that >95% of the cells were ASM cells in several
randomly chosen perspectives.
Mab suppresses the increase of
intracellular Ca2+ induced by Ach
Fluorescent intensity of Ca2+
determined with the Varioskan Flash
As shown in Fig. 5, Ach
(10−4 M) significantly increases Ca2+
fluorescent intensity when it was determined with the multimode
microplate reader. Mab (10−3, 10−4,
10−5, 10−6 and 10−7 mmol/l)
significantly suppressed this increase in a concentration-dependent
manner. The inhibitory rates of intracellular Ca2+ at
different concentrations of Mab, from low to high, were 14.93,
24.73, 40.06, 48.54 and 57.13%, respectively (Fig. 5).
Fluorescent intensity of
Ca2+ observed under the inverted system microscope
Representative images of Ca2+
fluorescence obtained from the inverted fluorescent microscope are
indicated in Fig. 6. More fluorescent
spots and higher fluorescent intensity can be observed in the
sample treated with Ach (Fig. 6A)
compared with those in the samples pre-incubated with
10−3, 10−4, 10−5, 10−6
and 10−7 mmol/l of Mab and subsequently with Ach
(Fig. 6B–F). The fluorescent intensity
of the sample treated with the highest concentration of Mab,
10−3 mmol/l (Fig. 6B), was
the least among all the Mab-treated samples (Fig. 6B–F).
Intracellular Ca2+ levels
determined with immunocytometry systems
The geometric mean (Geo Mean) of the M1 range in the
diagram of flow cytometry (Fig. 7) was
analyzed to determine Ca2+ fluorescent intensity. Ach
(10−4 M) significantly increases intracellular
Ca2+ compared with the control and Mab at the
concentration of 10−3 mmol/l, and 10−4 mmol/l
significantly suppresses the elevation of Ca2+
fluorescent intensity induced by Ach. When assessing the peak in
Fig. 7C, the peak of the cells loaded
with Fluo-3/AM following treatment with the highest concentration
of Mab, 10−3 mmol/l, shifts significantly to the left
compared with that in Fig. 7B (treated
with Ach 10−4 M alone). Geo Mean in the flow cytometry
diagram of the cells treated with Mab decreases in a
concentration-dependent manner, as shown from Fig. 7G to Fig.
7C.
 | Figure 7.Geometric mean (Geo Mean) of range M1
in the column figure, calculated based on the diagrams of the
immunocytometry systems. The cells loaded with Fluo-3/AM were
respectively treated with 10−7, 10−6,
10−5, 10−4 and 10−3 mmol/l of
mabuterol hydrochloride (Mab), and subsequently, their range of M1
in the flow cytometry diagram was decreased in a
concentration-dependent manner as shown in (G), (F), (E), (D) and
(C). The diagram in (B) illustrates the cells treated with
10−4 M acetylcholine (Ach) alone and (A) without
treatment. Data are expressed as mean ± standard error of the mean
obtained from three independent experiments. *P<0.05 and
**P<0.01 compared to the group treated with Ach, and
###P<0.001 compared to the control by analysis of
variance followed by least significant difference using SPSS
16.0. |
Discussion
ASM cells are involved in the pathophysiology of
numerous airway diseases, such as airway remodeling and
intracellular calcium overload (15).
The cell has become of interest in the study of the mechanisms of
bronchial asthma and chronic obstructive pulmonary disease.
Currently, two methods, i.e. with and without
enzymes to digest ASM pieces, are commonly used to prepare primary
ASM cells. It is known that the former may obtain the cells in a
short period of time (16). However,
it may lead to a less successful rate of the cell preparation, as
the ASM cells are extremely susceptible to injury from physical or
chemical factors. The reasons include that it is difficult to
control the exact quantity of enzymes and digestion time in
addition to the vulnerability of the cells. By contrast, not using
enzymes is relatively simple to handle and the step is mild for the
cells, but it is difficult for the cells to migrate out of the
tissue block due to its untreated toughness, and therefore, the
culture time is extended (17). An
improved method without CPTT was discussed in a previous study
(11). In this study, the culture
method with CPTT was tested and various periods of time, i.e. 5,
10, 20, 30 and 40 min, were assessed respectively to examine which
was the optimum time for enzyme digestion; 20 min was the best
treatment time. The time for the ASM cells to migrate out of the
tissue block was not fast compared with that of the method without
CPTT, as the time for digestion was not long enough. By contrast,
tissue blocks may become cotton- and wool-like and too many cells
were subjected to the enzymatic digestion if the time was too long.
Tissues digested after an appropriate duration in the present study
became softer and less tough, which caused the easier and earlier
migration of the cells from the tissue blocks. Additionally, tissue
blocks treated with collagenase can be repeatedly used ≤3 times.
Through trial and error experimental conditions, the cell culture
method established in the study is rapid, simple, efficient and
reproducible, which may provide a good platform for in vitro
studies in this research area.
The function of ASM cells is clearly regulated by
various signaling molecules. Activation of enzymes, protein
phosphorylation and release of calcium pools are all involved in
the transduction of the signaling molecules. Among them,
Ca2+ may play a central role and free Ca2+ in
the cytosol of ASM cells acts as a crucial secondary messenger in
numerous biological processes, such as contraction, proliferation,
gene transcription and secretion of signal mediators (4). Ach-induced Ca2+ transients and
oscillations in ASM cells have been previously studied and reported
(18,19). The present study identified that Mab
significantly suppressed the increased level of intracellular
Ca2+ induced by Ach.
The drug is a selective long-acting
β2-receptor agonist to be clinically used for asthma
treatment (20). In the present study,
to evaluate the mechanism of the antiasthmatic effect of Mab,
several methods of Ca2+ measurement were used, including
quantitative and qualitative analysis. Two calcium fluorescent
probes (Fura-2/AM and Fluo-3/AM) and three detection methods were
applied to determine Ca2+ fluorescent intensity. Images
of Ca2+ fluorescence in Fig.
6 illustrated the suppressive effect of Mab on the increased
Ca2+, which provided the information regarding the
drug's action though the measurement with the inverted fluorescent
microscope, as a method of qualitative analysis. Additionally,
Ca2+ fluorescent intensity was quantified through
single-wavelength detection with a multimode microplate reader. As
is illustrated in Fig. 5, Mab
concentration-dependently inhibits intracellular Ca2+,
which is clearly exhibited by the calcium inhibition rates. In
addition, the high concentration of Mab (10−3 and
10−4 mmol/l) significantly suppressed the elevation of
Ca2+ fluorescent intensity induced by Ach, which was
obtained from the Geo Mean of range M1 in the flow cytometry
diagram (Fig. 7). A total of 10,000
cells in each group were automatically captured and analyzed with
the equipment so as to compare the data of the groups. Similar
conclusions arose with these detective methods. Mab significantly
inhibits the Ca2+ increase induced by Ach in guinea pig
ASM cells.
Ach binds G-protein coupled receptors on the
membrane of ASM cells to activate phospholipase C, and
subsequently, inositoltrisphosphate (IP3) is produced under its
catalysis (21). In addition, Ach can
stimulate cluster of differentiation 38 to generate cADP-Ribose
(cADPR). Ca2+ is released from SR into the cytoplasm
subsequent to IP3 stimulating the clusters of IP3 receptors (IP3Rs)
on the membrane of SR and/or cADPR stimulating the ryanodine
receptor (RyR) (Fig. 8) (22). There is a possibility of RyR increasing
again via the mechanism of calcium-induced calcium release
following the activation of IP3R when the level of intracellular
Ca2+ is high enough, which leads to the evacuation of
the SR store and the accumulation of cytoplasmic Ca2+
(23). Intracellular Ca2+
accumulation eventually results in ASM contraction, proliferation
and migration. Our previous study identified that the mechanism of
tradinterol suppression on the elevation of intracellular
Ca2+ may be involved in the IP3R pathway (11). As was suggested in previous studies,
tradinterol is a new type of long-acting β2-agonist
(24,25). In the present study, Mab was proved to
significantly inhibit the calcium increase induced by Ach in guinea
pig ASM cells. Further investigation of whether the suppressive
activity of the drug on the calcium is the result of the
interaction between IP3R and RyR signaling pathways (Fig. 8) is required.
In conclusion, the renewed method of ASM cell
culture has successfully been proved. Additionally, it is clearly
shown that Mab significantly suppresses the increased level of
intracellular Ca2+ induced by Ach through three
measurement methods with a specific fluorescent probe in the ASM
cells. Due to the mechanism of calcium increase induced with Ach
and the suppressive effect of Mab on the increased level of
intracellular Ca2+, more studies should be performed to
clarify the mechanism of the suppression in detail, in which RyR
and/or the IP3R signaling pathway may provide innovative ideas with
further research.
References
|
1
|
James A, Mauad T, Abramson M and Green F:
Airway smooth muscle hypertrophy and hyperplasia in asthma. Am J
Respir Crit Care Med. 186:568–569. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siddiqui S, Redhu NS, Ojo OO, Liu B,
Irechukwu N, Billington C, Janssen L and Moir LM: Emerging airway
smooth muscle targets to treat asthma. Pulm Pharmacol Ther.
26:132–144. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Noble PB, Pascoe CD, Lan B, Ito S,
Kistemaker LE, Tatler AL, Pera T, Brook BS, Gosens R and West AR:
Airway smooth muscle in asthma: Linking contraction and
mechanotransduction to disease pathogenesis and remodelling. Pulm
Pharmacol Ther. 29:96–107. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Koopmans T, Anaparti V, CastroPiedras I,
Yarova P, Irechukwu N, Nelson C, PerezZoghbi J, Tan X, Ward JP and
Wright DB: Ca2 handling and sensitivity in airway smooth
muscle: Emerging concepts for mechanistic understanding and
therapeutic targeting. Pulm Pharmacol Ther. 29:108–120. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Boese M, Busse R, Mülsch A and
Schini-Kerth V: Effect of cyclic GMP-dependent vasodilators on the
expression of inducible nitric oxide synthase in vascular smooth
muscle cells: Role of cyclic AMP. Br J Pharmacol. 119:707–715.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dimitropoulou C, White RE, Ownby DR and
Catravas JD: Estrogen reduces carbachol-induced constriction of
asthmatic airways by stimulating large-conductance voltage and
calcium-dependent potassium channels. Am J Respir Cell Mol Biol.
32:239–247. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang IY, Bai Y, Sanderson MJ and Sneyd J:
A mathematical analysis of agonist- and KCl-induced Ca(2+)
oscillations in mouse airway smooth muscle cells. Biophys J.
98:1170–1181. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yamamoto H, Nagata M, Tabe K, Suzuki S,
Maruo H, Sakamoto Y, Yamamoto K and Dohi Y: The inhibitory effect
of long-acting beta-adrenergic agonists, mabuterol, clenbuterol and
fenoterol on ‘morning dipping’ in patients with asthma. Arerugi.
39:21–27. 1990.(In Japanese). PubMed/NCBI
|
|
9
|
Osada E, Murai T, Ishizaka Y and Sanai K:
Pharmacological studies of mabuterol, a new selective beta
2-stimulant. II: Effects on the cardiovascular system and smooth
muscle organs. Arzneimittelforschung. 34(11A): 1641–1651.
1984.PubMed/NCBI
|
|
10
|
Akahane K, Furukawa Y, Ogiwara Y, Haniuda
M and Chiba S: Beta-adrenoceptor blocking effects of a selective
beta 2-agonist, mabuterol, on the isolated, blood-perfused right
atrium of the dog. Br J Pharmacol. 97:709–716. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu J, Zhang Y, Li Q, Zhuang Q, Zhu X, Pan
L and Cheng M: An improved method for guinea pig airway smooth
muscle cell culture and the effect of SPFF on intracellular
calcium. Mol Med Rep. 10:1309–1314. 2014.PubMed/NCBI
|
|
12
|
Mosmann T: Rapid colorimetric assay for
cellular growth and survival: Application to proliferation and
cytotoxicity assays. J Immunol Methods. 65:55–63. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Orlandi A, Calzetta L, Doldo E, Tarquini
C, Matera MG and Passeri D: Brain natriuretic peptide modulates
calcium homeostasis and epidermal growth factor receptor gene
signalling in asthmatic airways smooth muscle cells. Pulm Pharmacol
Ther. 31:51–54. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu B, Yang J, Wen Q and Li Y:
Isoliquiritigenin, a flavonoid from licorice, relaxes guinea-pig
tracheal smooth muscle in vitro and in vivo: Role of cGMP/PKG
pathway. Eur J Pharmacol. 587:257–266. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pelaia G, Renda T, Gallelli L, Vatrella A,
Busceti MT, Agati S, Caputi M, Cazzola M, Maselli R and Marsico SA:
Molecular mechanisms underlying airway smooth muscle contraction
and proliferation: Implications for asthma. Respir Med.
102:1173–1181. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yamakage M, Hirshman CA and Croxton TL:
Volatile anesthetics inhibit voltage-dependent Ca2
channels in porcine tracheal smooth muscle cells. Am J Physiol.
268:L187–L191. 1995.PubMed/NCBI
|
|
17
|
Wu BN, Lin RJ, Lo YC, Shen KP, Wang CC,
Lin YT and Chen IJ: KMUP-1, a xanthine derivative, induces
relaxation of guinea-pig isolated trachea: The role of the
epithelium, cyclic nucleotides and K channels. Br J Pharmacol.
142:1105–1114. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bergner A and Sanderson MJ:
Acetylcholine-induced calcium signaling and contraction of airway
smooth muscle cells in lung slices. J Gen Physiol. 119:187–198.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Perez JF and Sanderson MJ: The frequency
of calcium oscillations induced by 5-HT, ACH and KCl determine the
contraction of smooth muscle cells of intrapulmonary bronchioles. J
Gen Physiol. 125:535–553. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kawakami Y: First clinical studies on
mabuterol. A summarizing report. Arzneimittelforschung. 34(11A):
1699–1700. 1984.PubMed/NCBI
|
|
21
|
Gosens R, Zaagsma J, Grootte Bromhaar M,
Nelemans A and Meurs H: Acetylcholine: A novel regulator of airway
smooth muscle remodelling? Eur J Pharmacol. 500:193–201. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jude JA, Wylam ME, Walseth TF and Kannan
MS: Calcium signaling in airway smooth muscle. Proc Am Thorac Soc.
5:15–22. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mahn K, Ojo OO, Chadwick G, Aaronson PI,
Ward JP and Lee TH: Ca(2+) homeostasis and structural and
functional remodelling of airway smooth muscle in asthma. Thorax.
65:547–552. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gan LL, Wang MW, Cheng MS and Pan L:
Trachea relaxing effects and beta2-selectivity of SPFF, a newly
developed bronchodilating agent, in guinea pigs and rabbits. Biol
Pharm Bull. 26:323–328. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hao Z, Zhang Y, Pan L, Su X, Cheng M, Wang
M, Zhao H and Wu Y: Comparison of enantiomers of SPFF, a novel
beta2-Adrenoceptor agonist, in bronchodilating effect in guinea
pigs. Biol Pharm Bull. 31:866–872. 2008. View Article : Google Scholar : PubMed/NCBI
|