Introduction
The purinergic 2X7 receptor (P2X7R) is significantly
expressed in CD31+ endothelial cells and
CD68+ macrophages in atherosclerotic lesions in human
carotid arteries (1). P2X7R may
also modulate the release of several cytokines known to promote
atherosclerosis, including interleukin (IL)-1β, IL-6 and tumor
necrosis factor α through immune cells (2). Although this evidence suggests that
there is an association between P2X7R and the development of
atherosclerosis, the mechanisms through which P2X7R promotes the
production of mature IL-1β by macrophages by modulating the
nucleotide-binding oligomerization domain-like receptor protein 3
(NLRP3) inflammasome remain unknown.
P2X7R has attracted the most attention out of all
the purinergic receptor family members, as it is involved in the
inflammatory response by modulating the production of cytokines
(such as IL-1β) by immune cells (2). However, the caspase-1
hydrolysis-dependent proIL-1β maturation also relies on NLRP3
inflammasome activity (7,8).
NLRP3 inflammasomes are known as cytoplasmic pattern
recognition receptors (PRRs) (3,4).
NLRP3 interacts with the adaptor molecule ASC (also known as PYD
and CARD domain containing PYCARD) and caspase-1 to form a large
multiprotein complex known as the NLRP3 inflammasome. Pro-caspase-1
is then converted into active caspase-1, which hydrolyzes proIL-1β
into active IL-1β (5). The NLRP3
inflammasome can be activated by a wide range of stimuli, including
intracellular cholesterol crystals (6,7),
uric acid crystals (8,9), extracellular amyloids (10,11), adenosine triphosphate (ATP)
(9,12,13), silicon dioxide and aluminum salts
(14,15). A number of extracellular
substances activate the NLRP3 inflammasome through P2X7R located on
the plasma membrane (16,17).
In vitro studies have demonstrated that
cholesterol crystals can activate the NLRP3 inflammasome and
promote the release of IL-1β by macrophages with the assistance of
lipopolysaccharides (LPS) (7,8).
This is mainly due to the fact that IL-1β production involves a
complex process and requires two independent signals (18,19). The first signal induces IL-1β mRNA
expression through the Toll-like receptor 4 (TLR4) pathway, which
is required for proIL-1β synthesis. The second signal is the
lysozyme-dependent NLRP3 inflammasome activation and active
caspase-1 production (8). Unlike
LPS, cholesterol crystals cannot activate TLR4 signaling.
Therefore, cholesterol crystals can only induce IL-1β production
with the assistance of LPS (7,8).
However, to the best of our knowledge, at present, evidence proving
the high level of LPS in the plasma of patients with coronary heart
disease is insufficient.
Thus, in addition to cholesterol crystals, there may
be other endogenous substances activating the NLRP3 inflammasome in
patients with atherosclerosis, such as oxidized low-density
lipoprotein (oxLDL). Unlike cholesterol crystals, oxLDL has a
function similar to LPS (20).
oxLDL can promote the release of IL-1β through direct TLR4
activation independent of LPS (21–25). Therefore, it is worth
investigating whether extracellular oxLDL activates the NLRP3
inflammasome through the upregulation of P2X7R in macrophages in a
manner simlilar to amyloid A and ATP.
In this study, we analyzed the expression and
distribution of P2X7R and NLRP3 in human coronary plaque. In order
to understand the role of P2X7R in modulating NLRP3 inflammasome
activity, we investigated the mechanisms behind the P2X7R-mediated
NLRP3 inflammasome activation induced by oxLDL in vitro. We
also examined the role of P2X7R in the progression of
atherosclerosis in apoE−/− mice. This study was approved
by the Research Ethics Committee of the Second Affiliated Hospital
of Dalian Medical University and the Animal Care and Use Committee
of Dalian Medical University, Dalian, China.
Materials and methods
Antibodies and reagents
The anti-IL-1β and anti-NLRP3 antibodies used in the
experiments in the present study were purchased from Proteintech
Group (Chicago, IL, USA). Anti-caspase-1 and anti-CD68 antibodies
were purchased from Wuhan Boster Biological Technology, Ltd.
(Wuhan, China). Anti-P2X7 antibody was from Beijing Bioss
Biosynthesis Biotechnology Co. Ltd. (Beijing, China). Anti-protein
kinase R (PKR) (phospho T446) antibody was from Abcam (Cambridge,
MA, USA). Short interfering RNA (siRNA) and negative control (NC)
siRNA were purchased from Guangzhou RiboBio Co., Ltd. (Guangzhou,
China).
Cell preparation
THP-1, a human monocyte cell line, was obtained from
the American Type Culture Collection (ATCC, Manassas, VA, USA). The
cells were incubated in RPMI-1640 with 10% (v/v) heat-inactivated
fetal bovine serum (FBS; Thermo Scientific HyClone, Logan, UT,
USA), 100 U/ml penicillin and 100 g/ml streptomycin
(Invitrogen/Life Technologies, Carlsbad, CA, USA). Phorbol
12-myristate 13-acetate (PMA; Sigma-Aldrich, St. Louis, MO, USA)
was used to induce the differentiation of the THP-1 cells into
macrophages and the cells were then cultured at 37°C in a
humidified atmosphere of 5% CO2 in room air.
Human coronary artery biopsy
preparation
The paraffin section of the right coronary artery
and the anterior descending branch of the left coronary artery from
an autopsy specimen were provided by the Department of Pathology,
University of South China, Hengyang, China.
LDL isolation and modification
Native human LDL (nLDL) was isolated from plasma by
ultracentrifugation. The density of the plasma was adjusted to 1.2
g/ml by the addition of solid potassium bromide (KBr). Following
ultracentrifugation at 41,500 × g for 4 h at 4°C, very low-density
lipoprotein was removed, 3.0 ml of the lower layer was transferred
to another tube, 2 ml KBr-NaCl (d=1.18 g/ml) was added and the
samples were gently mixed. The tubes were ultracentrifuged at
41,500 × g for 4 h at 4°C, and the LDL fraction was removed. The
LDL fraction was dialyzed against 0.5 mM NaCl, pH 7.4, for 24 h at
4°C to remove EDTA. LDL was oxidized using CuSO4 as
previously described (26).
Histological analysis
Frozen cross-sections (8 μm thick) of the
aortic sinus of apoE−/− mice and paraffin-embedded
cross-sections of human coronary artery (5 μm thick) were
stained with primary antibodies (P2X7R, CD68 and NLRP3) and
visualized with the appropriate secondary antibodies [from the
SABC-POD Rabbit IgG IHC kit (SA1022); Wuhan Boster Biological
Technology, Ltd.; goat anti-rabbit IgG, Cy3 conjugated (CW0159) and
FITC conjugated (CW0114) secondary antibodies for fluorescence were
from Cwbiotech, Beijing, China]. Conventional hematoxylin and eosin
(H&E) staining of the pathological sections was performed. Oil
Red O (kit provided by Nanjing Jiancheng Bioengineering Institute,
Nanjing, China) was manipulated with specification for observing
the cholesterol ester content in lipid-burdened cells and the
frozen animal aortic sections. Section images were captured using
Image-Pro Plus 6.0 software (Media Cybernetics, Inc., Rockville,
MD, USA) and analyzed using Adobe Photoshop CS5 software (Adobe
Systems Inc., San Jose, CA, USA).
Mice
Male apoE knockout (apoE−/−) mice (6
weeks old with a C57BL/6 background) were supplied by Charles River
Laboratories Inc. (Wilmington, MA, USA). For acclimation, the mice
were fed a rodent chow diet for 1 week before being fed a
high-cholesterol diet for 2, 4, 8 and 12 weeks (2% w/w cholesterol,
10% w/w lard). Mice in the control group were fed the rodent chow
diet for 1 week. The hearts were collected and fixed by perfusion
in situ with 10% neutralized formalin. The fixed hearts were
then placed in a freezer at −20°C for use as frozen serial
sections.
Enzyme-linked immunosorbent assay (ELISA)
for determining the levels of pro-inflammatory cytokines and
oxLDL
Cytokine levels in the cell supernatant and mouse
plasma were measured using ELISA kits (Neobioscience Technology
Co., Ltd., Beijing, China). ELISA kits for mouse plasma oxLDL
levels were from Cusabio Biotech Co., Ltd. (Wuhan, China).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the cells using the
E.Z.N.A.® HP Total RNA kit (Omega Bio-Tek, Inc.,
Norcross, GA, USA) following the instructions provided by the
manufacturer. The RNA samples were converted into cDNA using the
RevertAid First Strand cDNA Synthesis kit (Thermo Scientific
Fermentas, Waltham, MA, USA). Quantitative PCR was carried out
using the following primers: GAPDH sense, 5′-ATGACATCAAGAGGTGGTG-3′
and antisense, 5′-CATACCAGGAAATGAGCTTG-3′, annealing 60°C; NLRP3
sense, 5′-GTGTTTCGAAATCCCACTGTG-3′ and antisense,
5′-TCTGCTTCTCACGTACTTTCTG-3′, annealing 60°C and 57°C; P2X7R sense,
5′-GAACAATATCGACTTCCCCGG-3′ and antisense,
5′-TTATCGCCTGTTTCTCGGAAG3′, annealing 60°C and 55°C; IL-1β sense,
5′-CCTCCATTGATCATCTGTCTCTG3′ and antisense,
5′-GCTTGGATGTTTTAGAGGTTTCAG-3′, annealing 58°C.
Western blot analysis and
immunoprecipitation
The cells were suspended in the appropriate lysis
buffer. Following centrifugation (10,000 rpm for 10 min), the
protein content in the supernatants was quantified using the BCA
protein assay kit (Beijing ComWin Biotech Co., Ltd., Beijing,
China). The boiling denatured proteins were segregated by sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred onto PVDF membranes (Millipore, Billerica, MA, USA) at
4°C. The membranes carrying proteins were incubated with primary
antibody and detected with the appropriate secondary polyclonal
antibody by BeyoECL Plus (Beyotime Institute of Biotechnology,
Jiangsu, China). Antibodies against PKR and NLRP3 were used to
precipitate proteins from cell lysis in the presence of 20
μl protein A/G beads (Santa Cruz Biotechnology, Santa Cruz,
CA, USA) overnight at 4°C. Protein complexes were washed 4 times
with lysis buffer, and then incubated at 95°C for 5 min and
resolved by western blot analysis.
siRNA knockdown and transfection of cells
and injection of siRNA into mice
The THP-1 macrophages were transfected with NLRP3
siRNA, P2X7R siRNA, or the negative control (NC) siRNA at 50 nM
using the HiPerFect Transfection Reagent (Qiagen, Hilden, Germany).
In the animal experiments, a lentivirus carrying P2X7R siRNA
(GeneChem, Co., Ltd., Shanghai, China) was intravenously injected
into tail veins of the apoE−/− mice once every 2 weeks.
The apoE−/− mice in the negative control (NC) group were
injected with a lentiviral vector carrying scrambled siRNA. Mice
not injected with siRNA were used as the controls.
Statistical analysis
Data are presented as the means ± standard deviation
(SD). An unpaired two-tailed Student’s t-test was used for
comparisons between 2 groups. Statistical analysis for 3 or more
groups was performed using one-way ANOVA with the Tukey’s post-hoc
test. A value of P<0.05 was considered to indicate a
statistically significant differencde. Statistical analysis was
performed using IBM SPSS statistical software (version 21.0).
Results
P2X7R and NLRP3 expression levels in
atherosclerotic lesions in human coronary arteries
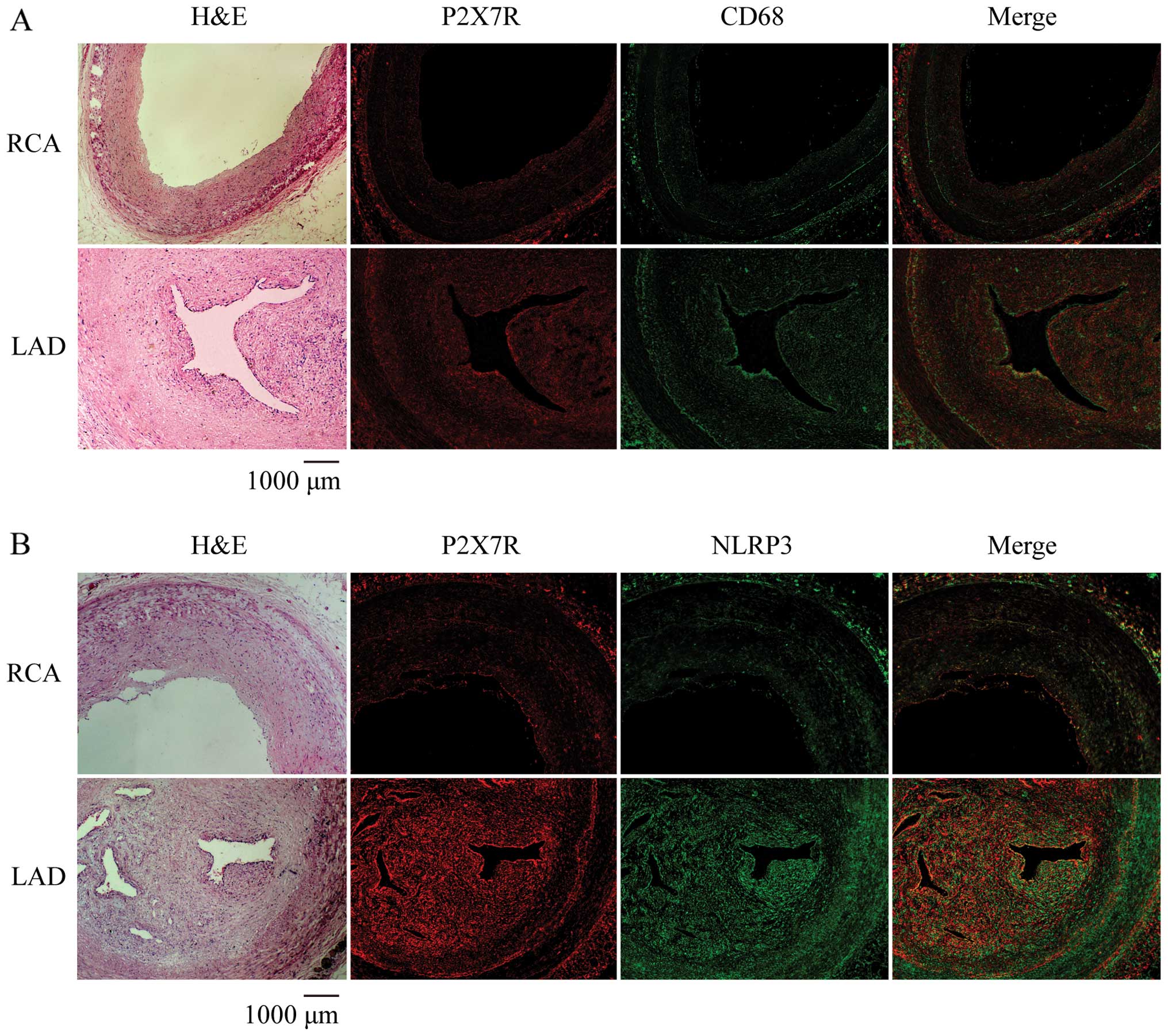
The morphological characteristics of the left
anterior descending coronary artery and right coronary artery from
the autopsy specimen was observed by H&E staining. The left
anterior descending artery showed apparent eccentric stenosis with
large amounts of foam cells compared to the right coronary artery
(Fig. 1). Immunofluorescence
staining for P2X7R, NLRP3 and the macrophage marker, CD68, using
specific antibodies revealed the co-expression of P2X7R and NLRP3
in the atherosclerotic plaques (Fig.
1B) with large numbers of macrophages (Fig. 1A). This suggests that the high
expression of P2X7R and NLRP3 in macrophages may be involved in the
progression of atherosclerosis.
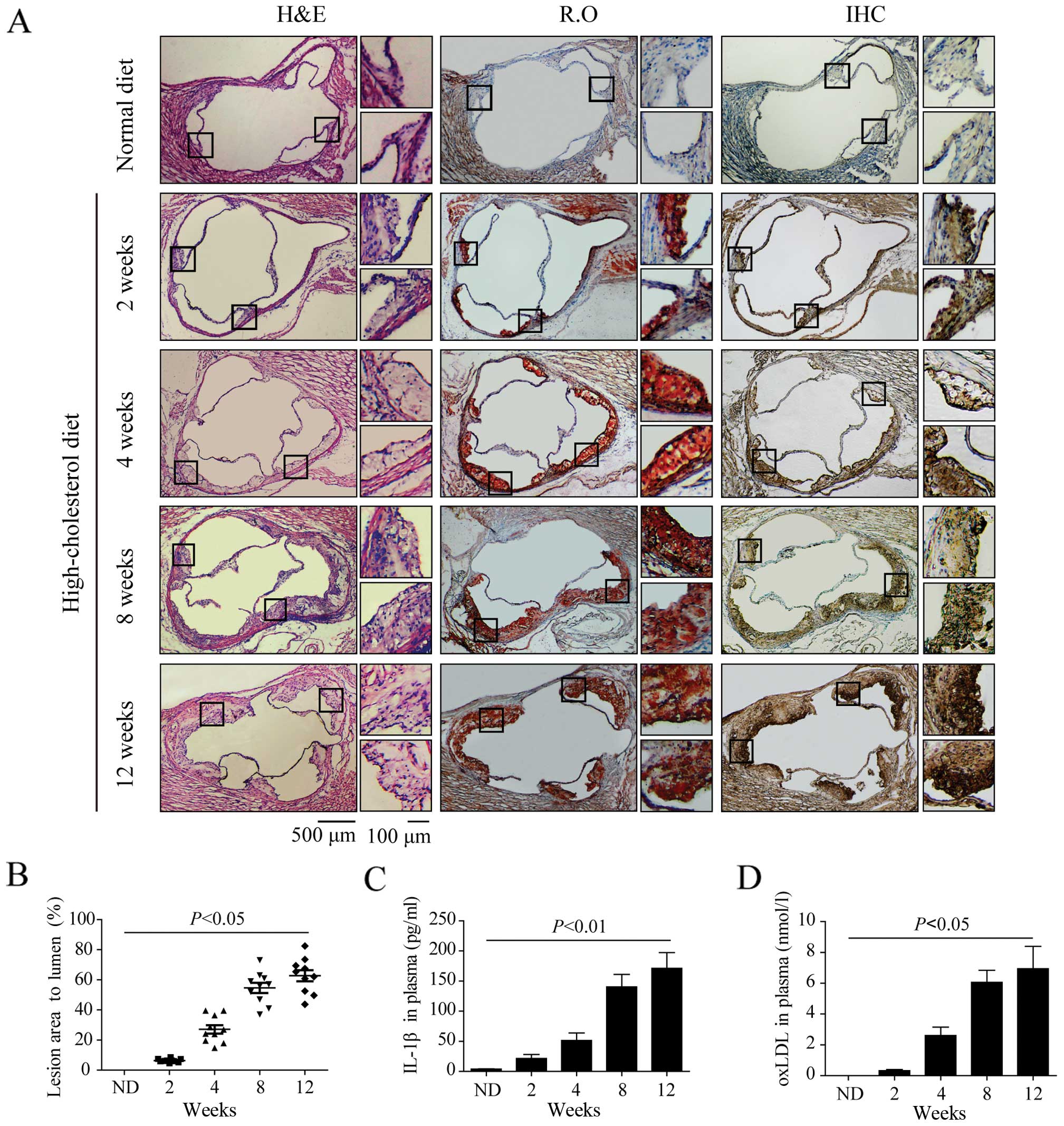
Increased P2X7R expression in
atherosclerotic lesions in apoE−/− mice
In this study, lesion size was determined by the
percentage of atherosclerotic lesions compared to the entire
cross-sectional vessel lumen area stained with H&E. There was
only a small amount of foam cells in the aortic sinuses of
apoE−/− mice fed a high-cholesterol diet for 2 weeks,
but P2X7R expression in the atherosclerotic lesions was
significantly increased (Fig. 2A)
compared with the control group (on a chow diet for 1 week). After
4 weeks on a high-cholesterol diet, more foam cells accumulated on
the arterial wall with larger lesion areas compared to those at 2
weeks (Fig. 2A and B). Oil Red O
staining and immunohistochemistry revealed that P2X7R was present
in the foam cell-rich areas (Fig.
2A). Atherosclerotic plaque formed in the aortic sinuses of the
apoE−/− mice after 8 and 12 weeks on a high-cholesterol
diet, and the lesion were significantly elevated toward the intimal
surface with cholesterol crystals. There were many newly formed
foam cells which hade accumulated in the lesions close to the
lumen. P2X7R was highly expressed in the atherosclerotic plaques,
particularly in the newly formed foam cells (Fig. 2A). ELISA revealed that the plasma
IL-1β and oxLDL levels in the apoE−/− mice fed a
high-cholesterol diet for 4, 8 and 12 weeks were significantly
higher than those of the mice in the control group (Fig. 2C and D).
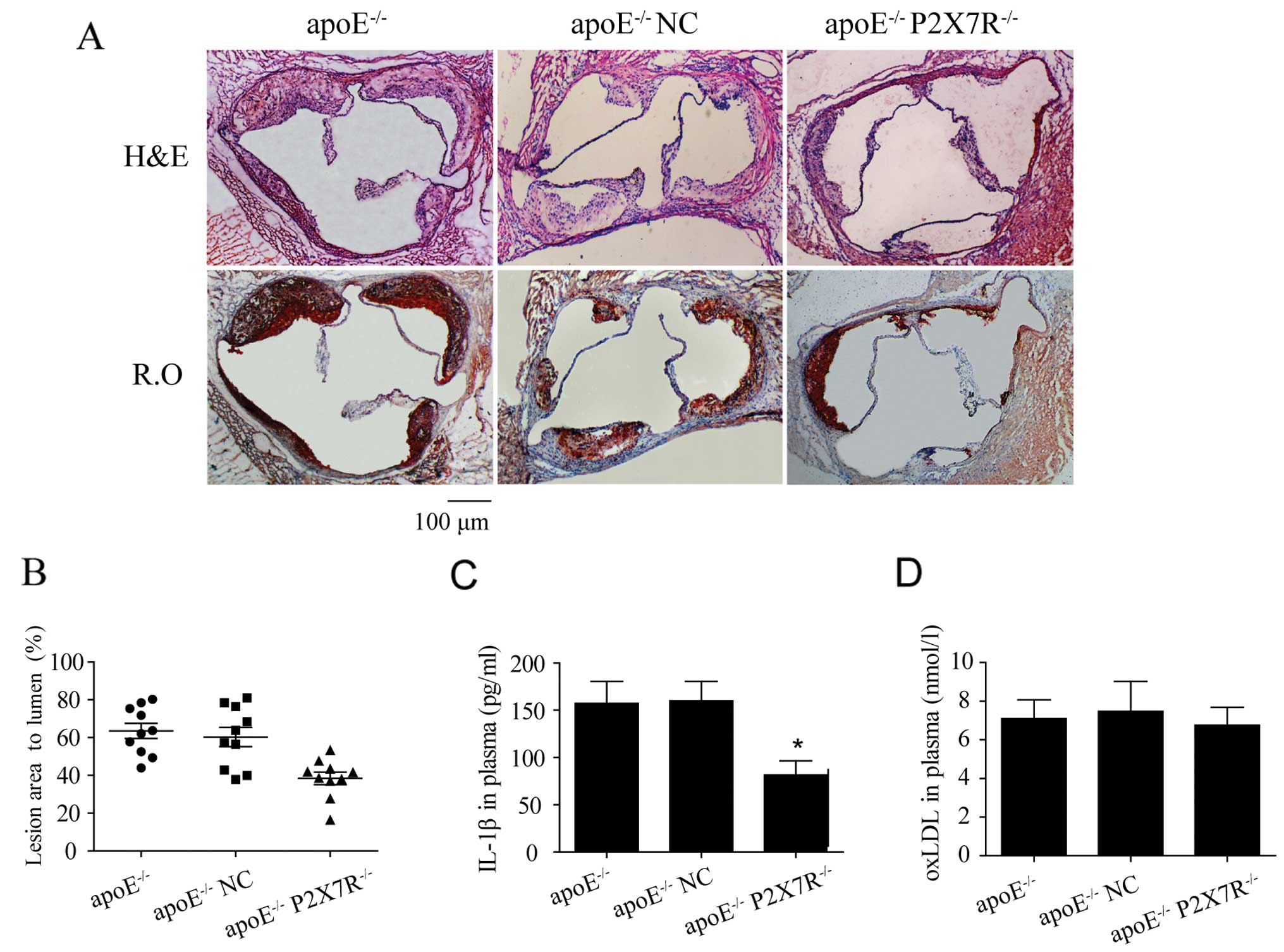
Effect of P2X7R on the formation of
atherosclerotic lesions in apoE−/− mice
To suppress the function of P2X7R, a lentiviral
vector carrying P2X7R siRNA was delivered to the apoE−/−
mice by a tail vein injection. The apoE−/− mice in the
negative control group were injected with a lentiviral vector
carrying scrambled siRNA and the mice in the control group were not
injected with siRNA. After 12 weeks on a high-cholesterol diet, the
aortic sinus plaque areas in the apoE−/− mice injected
with P2X7R siRNA (apoE−/− P2X7R−/−) were
significantly smaller than those in the mice in the other 2 groups
(Fig. 3A and B). More
importantly, in addition to many foam cells, the lesions in the
apoE−/− mice and apoE−/− NC mice exhibited
significant extracellular lipid accumulation, necrotic tissue and
some cholesterol crystals (Fig.
3A), indicating the formation of atherosclerotic plaque in the
aortic sinuses. However, the aortic sinus lesions in the
apoE−/− P2X7R−/− mice were mainly formed by
foam cell accumulation, which is consistent with the early
pathological characteristics of fatty streaks (Fig. 3A). However, a significantly
delayed progression of atherosclerosis was observed in the
apoE−/− P2X7R−/− mice (Fig. 3A and B).
Additionally, the plasma oxLDL levels were the same
in each group, which indicates that the P2X7R knockdown did not
affect the oxLDL plasma concentrations (Fig. 3D). However, the IL-1β levels in
the plasma of apoE−/− P2X7R−/− mice were
lower than those observed in the other 2 groups (Fig. 3C). This suggests that the
development of atherosclerosis mediated by P2X7R in mice may be
associated with the modulation of IL-1β activation and release.
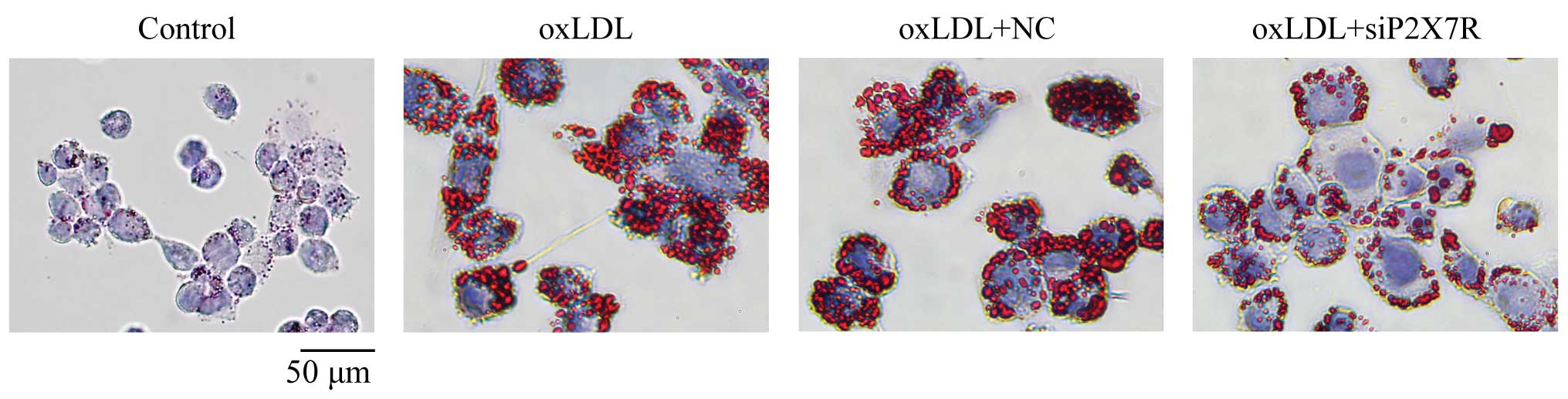
P2X7R affects foam cell formation
Oil Red O staining revealed that many lipid droplets
had accumulated in the THP-1 macrophages after 24 h of incubation
with 100 μg/ml oxLDL (Table
I and Fig. 4). Additionally,
the cytoplasmic cholesterol ester content accounted for >50% of
the total cholesterol. This indicates foam cell formation. There
were significantly fewer cholesterol esters (Table I) and lipid droplets (Fig. 4) in the P2X7R siRNA-treated
macrophages compared to the negative controls, suggesting that
P2X7R affects lipid loading in macrophages and foam cell
formation.
 | Table ICholesterol and cholesterol esters in
THP-1 macrophages (mg/g, n=3). |
Table I
Cholesterol and cholesterol esters in
THP-1 macrophages (mg/g, n=3).
| Group | TC | FC | CE | CE/TC (%) |
|---|
| Control | 194.2±16.7 | 143.4±12.1 | 51.1±4.3 | 26.3 |
| oxLDL | 464.8±13.5 | 188.2±17.7 | 276.3±15.8a | 59.4 |
| oxLDL + NC | 459.1±12.2 | 175.5±9.3 | 284.6±19.2a | 61.8 |
| oxLDL +
siP2X7R | 282.4±19.1 | 174.7±7.3 | 107.8±11.7b | 37.9 |
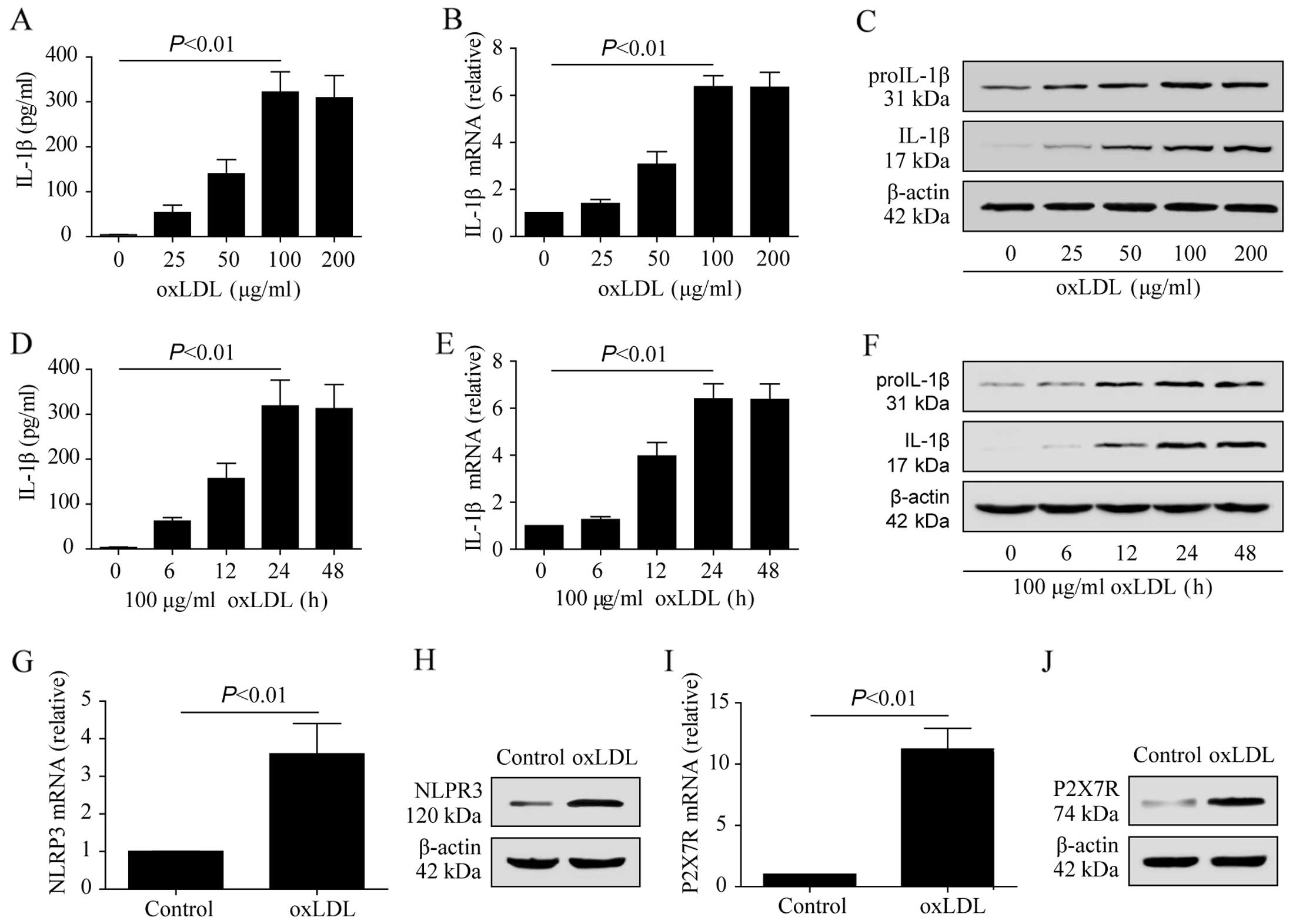
Regulatory role of oxLDL in the induction
of P2X7R, NLRP3 and IL-1β expression in macrophages
To demonstrate that oxLDL upregulates P2X7R and
NLRP3 in THP-1 macrophages and promotes the secretion of IL-1β, the
THP-1 macrophages were stimulated with 25, 50, 100 and 200
μg/ml oxLDL for 24 h. The IL-1β concentrations were
significantly higher in the oxLDL-treated groups compared to the
control group (Fig. 5A). The
cytoplasmic proIL-1β mRNA and protein levels, as well as the mature
IL-1β protein levels, were increased in the THP-1 macrophages
treated with oxLDL (Fig. 5B and
C). A concentration of 100 μg/ml of oxLDL was selected
to treat the THP-1 macrophages for 6, 12, 24 and 48 h to examine
the IL-1β expression and secretion. Following treatment with 100
μg/ml oxLDL for 24 h, the expression of mature IL-1β and
proIL-1β increased compared to the control group (Fig. 5E and F). IL-1β medium
concentration was also significantly increased (Fig. 5D). These results indicated that
oxLDL upregulated IL-1β expression in THP-1 macrophages and
promoted proIL-1β hydrolysis to activate IL-1β. oxLDL also
upregulated the the mRNA and protein expression levels of P2X7R and
NLRP3 in the THP-1 macrophages (Fig.
5G–J).
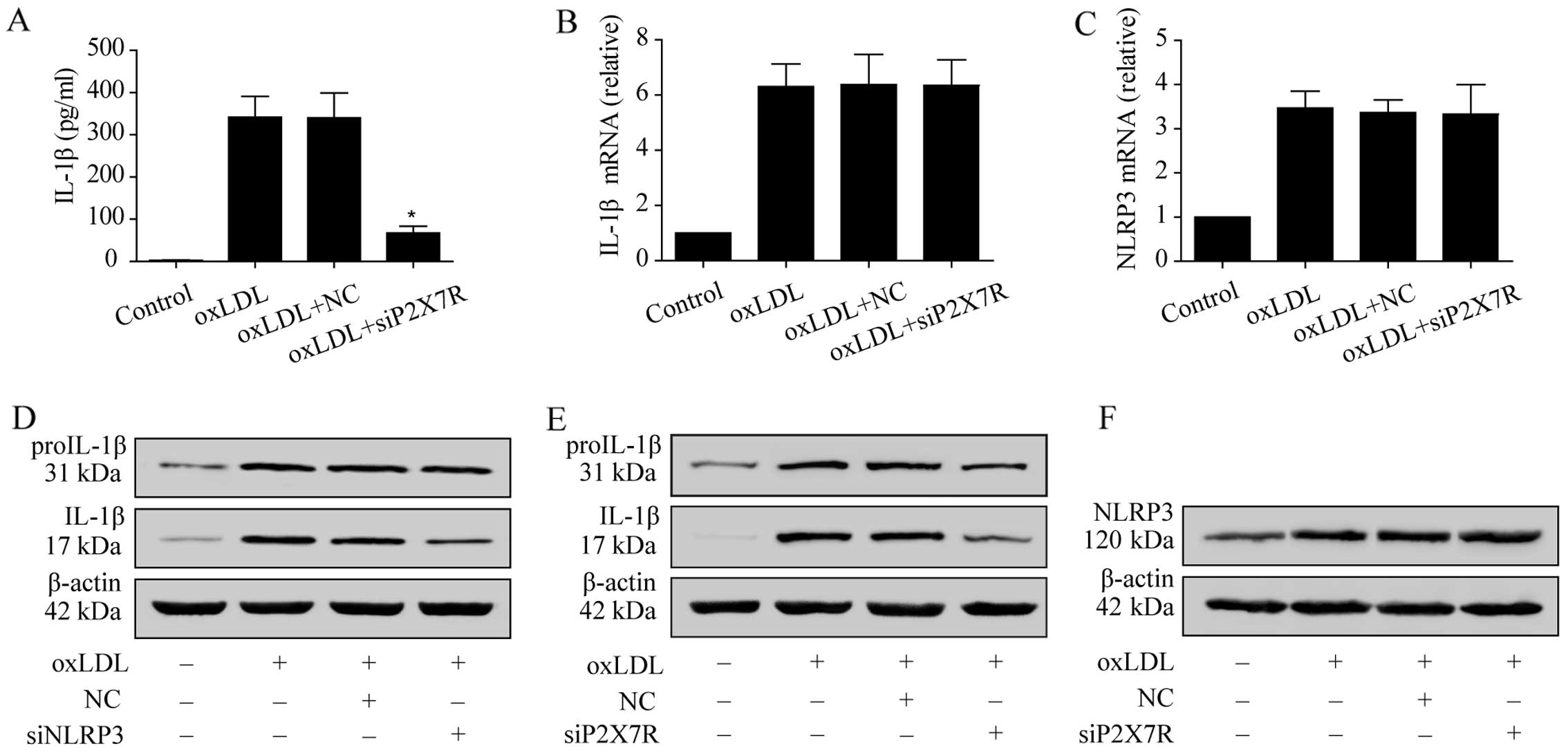
Regulatory role of P2X7R and the NLRP3
inflammasome in the production of IL-1β in macrophages
The IL-1β mRNA and proIL-1β protein expression
levels in the oxLDL-stimulated macrophages did not differ between
the negative control group and the P2X7R siRNA-treated group
(Fig. 6B and E). However,
proIL-1β activity was significantly suppressed (Fig. 6E). The concentration of IL-1β in
the medium was also reduced due to the suppression of P2X7R
(Fig. 6A), since proIL-1β
hydrolysis to mature IL-1β is controlled by NLRP3 inflammasome
activation. After NLRP3 expression was suppressed using siRNA,
mature IL-1β expression in the oxLDL-stimulated macrophages was
significantly lower than that in the negative controls, while
proIL-1β expression was unaltered (Fig. 6D). However, NLRP3 expression in
the macrophages in which P2X7R was knocked down following oxLDL
stimulation did not differ from that observed in the negative
control group (Fig. 6C and F).
These results indicate that, upon oxLDL stimulation, the expression
of P2X7R in macrophages regulates NLRP3 inflammasome function, but
not NLRP3 expression.
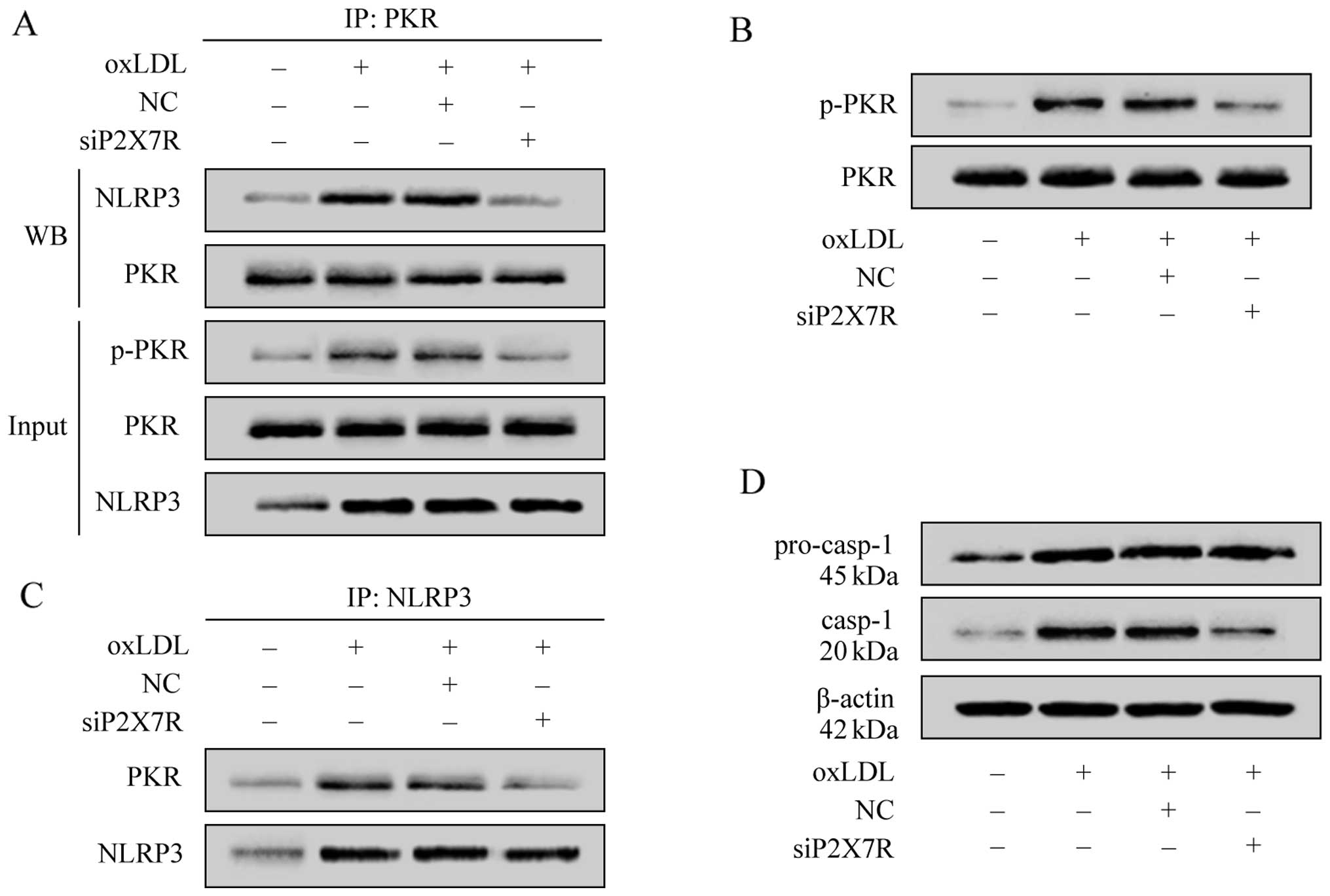
P2X7R regulates NLRP3 inflammasome
assembly by promoting PKR phosphorylation
To verify that P2X7R affects NLRP3 inflammasome
activity by regulating PKR phosphorylation upon stimulation with
oxLDL, we evaluated the phosphorylation level of PKR in the THP-1
macrophages using specific antibodies, and investigated the
interaction between phosphorylated PKR and NLRP3 using a
co-immunoprecipitation method. The results revealed that treatment
with oxLDL promoted PKR phosphorylation, upregulated NLRP3
expression (Fig. 7A–C) and
produced a large amount of active caspase-1 (Fig. 7D). The regulation of PKR
phosphorylation by oxLDL was significantly attenuated when P2X7R
was suppressed, which suggested that oxLDL promoted PKR
phos-phorylation by upregulating P2X7R. To examine the interaction
between PKR and NLRP3, we used a specific antibody to PKR in the
co-immunoprecipitation assay, which revealed that a large amount of
PKR formed a complex with NLRP3 after being phosphorylated by oxLDL
(Fig. 7A and C). After P2X7R was
suppressed by siRNA, the oxLDL-induced PKR phosphorylation was
significantly decreased. Consequently, there was a reduced
interaction between P2X7R and NLRP3 and a reduced caspase-1
activity (Fig. 7D). As the
production of active caspase-1 signals NLRP3 inflammasome assembly
and activation, these results indicate that oxLDL regulates NLRP3
inflammasome activity by upregulating P2X7R expression and
promoting PKR phosphorylation.
Discussion
P2X7R (27) and
NLRP3 (28) are abundant in
immune cells, such as mononuclear phagocytes, dendritic cells and T
cells. A number of substances activate the NLRP3 inflammasome
through the P2X7R pathway (29–31). Niemi et al (10) demonstrated that serum amyloid A
(SAA) induced proIL-1β expression and activated the inflammasome,
promoting macrophages to secrete mature IL-1β. The suppression of
caspase-1 and P2X7R hampered the production of mature IL-1β
triggered by SAA (10). Gicquel
et al (12) found that the
secretion of IL-1β by ATP-stimulated macrophages involved the
P2X7R/NLRP3 inflammasome pathway.
In the present study, in the plaque of the left
anterior descending coronary artery, P2X7R and NLRP3 were highly
co-expressed in areas with macrophage accumulation (Fig. 1). This suggests that the high
expression levels of P2X7R and NLRP3 in macrophages are involved in
the progression of atherosclerosis. This is supported by
Piscopiello et al (1), who
observed high P2X7R expression in CD31+ endothelial
cells and CD68+ macrophages in human carotid
atherosclerotic lesions.
To further illustrate the role of P2X7R in the
development of atherosclerosis, this study examined the expression
of P2X7R in the arterial walls of apoE−/− mice fed a
high cholesterol diet. We demonstrated that, in the atherosclerotic
lesions, the expression of P2X7R was high in the areas with foam
cell accumulation (Fig. 2A and
B). The plasma levels of IL-1β in mice with atherosclerosis
were also significantly higher than those observed in the control
group (Fig. 2C). However, P2X7R
expression detected in human coronary atherosclerotic plaque is not
sufficient to prove that P2XR7 is involved in the development of
atherosclerosis. It was thus necessary to observe the effects of
the knockdown of P2X7R on atherosclerosis. In vivo, the
knockdown of P2X7R in apoE−/− mice significantly delayed
the development of atherosclerosis (Fig. 3A and B) with lower plasma levels
of IL-1β (Fig. 3C). In the
apoE−/− mice injected with P2XR7 siRNA, the
atherosclerotic lesion area in the aortic root was smaller compared
to that in the negative control group, but foam cells were the main
component in lesions where cholesterol crystals and necrotic
material were not found.
These findings indicate that P2X7R, by modulating
IL-1β maturation, plays an important role in the development of
atherosclerosis. This is also supported by the finding of a
previous study demonstrating that apoE−/−
Casp−/− mice had suppressed plaque areas and macrophage
infiltration compared to apoE−/− mice (32). In our animal experiments, P2X7R
expression in foam cell-rich areas is particularly prominent;
therefore, in this study, we also examined the effects of P2X7R
expression on macrophage foam cell formation in vitro. After
the cells were incubated with oxLDL for 24 h, macrophage foam cell
formation was evident. P2X7R knockdown significantly delayed this
formation (Table I and Fig. 4). Although no studies have
suggested that P2X7R in macrophages directly mediates lipid
phagocytosis, P2X7R has been shown to promote foam cell formation
by modulating the NLRP3/IL-1β pathway (5,34).
Evidence from the present and previous studies
suggests that, in atherosclerosis, oxLDL is the most likely
candidate to upregulate P2X7R and activate the NLRP3 inflammasome.
Firstly, the present study showed that oxLDL promoted macrophage
foam cell formation, while macrophage foam cell formation was
suppressed by P2X7R knockdown. Moreover, in vivo, P2X7R
knockdown delayed the progression of atherosclerosis. Secondly,
immunohistochemical studies have revealed that the NLRP3
inflammasome is activated in plaque macrophages (19), while oxLDL modulates NLRP3
inflammasome activity in mouse macrophages and activates caspase-1
(34,35). Thirdly, a number of substances,
such as amyloid protein A (10,11) and ATP (9,12,13) have been shown to activate the
NLRP3 inflammasome by upregulating P2X7R expression. Finally,
patients with coronary heart disease exhibit higher plasma oxLDL
levels (36). Moreover, studies
have shown that patients with coronary artery disease do not
exhibit high plasma LPS concentrations, and that oxLDL differs from
cholesterol crystals (7,8) in that it can activate TLR4 without
LPS, while oxLDL itself also has a cholesterol component (21–25).
To verify our hypothesis, we incubated the THP-1
macrophages with oxLDL. oxLDL not only upregulated P2X7R, NLRP3 and
proIL-1β expression, but also promoted the production and release
of active IL-1β (Fig. 5A–J).
Masters et al (20) found
that oxLDL upregulated IL-1β mRNA expression and promoted the
release of IL-1β by mouse bone marrow-derived macrophages. These
conclusions are consistent with those of the present study. By
knocking down the expression of P2X7R and NLRP3 in macrophages
using specific siRNA, we demonstrated that the generation of mature
IL-1β upon oxLDL stimulation was dependent on P2X7R and the NLRP3
inflammasome (Fig. 6D and E).
Since proIL-1β maturation is directly controlled by the NLRP3
inflammasome under the effects of oxLDL, P2X7R either modulates
NLRP3 expression or the NLRP3 inflammasome assembly and
activation.
Our experiments revealed that, after the P2X7R
knockdown, proIL-1β maturation was evidently inhibited, but oxLDL
still significantly upregulated NLRP3 and proIL-1β expression. This
indicates that, upon oxLDL stimulation, P2X7R in macrophages
modulates proIL-1β maturation processes (Fig. 6A, B and E), but does not regulate
NLRP3 and proIL-1β expression (Fig.
6B, C, E and F). Therefore, P2X7R is likely to affect NLRP3
inflammasome activity by regulating its assembly. In the study by
Lu et al (37), it was
demonstrated that phosphorylated PKR, under the effects of ATP, is
essential for NLRP3 inflammasome assembly and activation. It should
be noted that, although ATP is considered a classic ligand to
P2X7R, these studies did not investigate the regulatory effects of
P2X7R on NLRP3 inflammasome activity. In this study, we
demonstrated that, upon oxLDL stimulation, P2X7R knockdown reduced
PKR phosphorylation and the amount of NLRP3 combined with PKR
(Fig. 7A and B). NLRP3
inflammasome-mediated caspase-1 (Fig.
7D) and mature IL-1β levels were also significantly reduced
(Fig. 6D). Therefore, P2X7R may
regulate NLRP3 inflammasome activity by regulating NLRP3
inflammasome assembly rather than affecting its expression.
In the development of atherosclerosis, cholesterol
may activate the NLRP3 inflammasome in two ways. One includes
cholesterol crystal formation following endocytosis through
receptors related to lipid metabolism, such as CD36 (21,38,39). These cholesterol crystals activate
the NLRP3 inflammasome through the cathepsin B-dependent
destabilizing lysosome (7,8).
The second way is by upregulating macrophage NLRP3 and IL-1β mRNA
and protein expression in the form of oxLDL through the TLR4/NF-κB
pathway (33). At the same time,
oxLDL initiates phosphorylated PKR interaction with NLRP3 by
upregulating P2X7 receptors, and thus promotes NLRP3 inflammasome
assembly and activation.
In conclusion, the present study demonstrates that
P2X7R and NLRP3 are expressed at hight levels in foam cell-rich
human coronary atherosclerotic lesion areas, and that P2X7R is
involved in the progression of atherosclerosis in vivo and
in vitro. Upon oxLDL stimulation, P2X7R upregulation
modulates NLRP3 inflammasome activation by enhancing PKR
phosphorylation, which promotes the production of mature IL-1β by
macrophages. This is possibly one of the mechanisms through which
P2X7R participates in the progression and development of
atherosclerosis.
Acknowledgments
This study was supported by a grant from the
National Natural Science Foundation of China (30670836).
References
|
1
|
Piscopiello M, Sessa M, Anzalone N, et al:
P2X7 receptor is expressed in human vessels and might play a role
in atherosclerosis. Int J Cardiol. 168:2863–2866. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mehta N, Kaur M, Singh M, et al:
Purinergic receptor P2X7: A novel target for anti-inflammatory
therapy. Bioorg Med Chem. 22:54–88. 2014. View Article : Google Scholar
|
|
3
|
Wang LL, Wang HY and Qu P: Role and
relationships of pattern-recognition receptors in atherosclerosis
(In Chinese). Chin J Arterioscler. 20:951–955. 2012.
|
|
4
|
Jia Y, Zhou L and Li XH: Advance in the
study of nod-like receptor protein-3 inflammasome and
atherosclerosis (In Chinese). Chin J Arterioscler. 22:79–84.
2014.
|
|
5
|
Yang CS, Shin DM and Jo EK: The role of
NLR-related protein 3 inflammasome in host defense and inflammatory
diseases. Int Neurourol J. 16:2–12. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Grebe A and Latz E: Cholesterol crystals
and inflammation. Curr Rheumatol Rep. 15:3132013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Duewell P, Kono H, Rayner KJ, et al: NLRP3
inflammasomes are required for atherogenesis and activated by
cholesterol crystals. Nature. 464:1357–1361. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rajamäki K, Lappalainen J, Oörni K, et al:
Cholesterol crystals activate the NLRP3 inflammasome in human
macrophages: a novel link between cholesterol metabolism and
inflammation. PLoS One. 5:e117652010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Riteau N, Baron L, Villeret B, et al: ATP
release and purinergic signaling: a common pathway for
particle-mediated inflammasome activation. Cell Death Dis.
3:e4032012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Niemi K, Teirilä L, Lappalainen J, et al:
Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor
and a cathepsin B-sensitive pathway. J Immunol. 186:6119–6128.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Eklund KK, Niemi K and Kovanen PT: Immune
functions of serum amyloid A. Crit Rev Immunol. 32:335–348. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gicquel T, Victoni T, Fautrel A, et al:
Involvement of purinergic receptors and NOD-like receptor-family
protein 3-inflammasome pathway in the adenosine
triphosphate-induced cytokine release from macrophages. Clin Exp
Pharmacol Physiol. 41:279–286. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gombault A, Baron L and Couillin I: ATP
release and purinergic signaling in NLRP3 inflammasome activation.
Front Immunol. 3:4142013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kuroda E, Ishii KJ, Uematsu S, et al:
Silica crystals and aluminum salts regulate the production of
prostaglandin in macrophages via NALP3 inflammasome-independent
mechanisms. Immunity. 34:514–526. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hornung V, Bauernfeind F, Halle A, et al:
Silica crystals and aluminum salts activate the NALP3 inflammasome
through phagosomal destabilization. Nat Immunol. 9:847–856. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tschopp J and Schroder K: NLRP3
inflammasome activation: The convergence of multiple signalling
pathways on ROS production? Nat Rev Immunol. 10:210–215. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Latz E, Xiao TS and Stutz A: Activation
and regulation of the inflammasomes. Nat Rev Immunol. 13:397–411.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wen H, Miao EA and Ting JP: Mechanisms of
NOD-like receptor-associated inflammasome activation. Immunity.
39:432–441. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
McGettrick AF and O’Neill LA: How
metabolism generates signals during innate immunity and
inflammation. J Biol Chem. 288:22893–22898. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Masters SL, Dunne A, Subramanian SL, et
al: Activation of the NLRP3 inflammasome by islet amyloid
polypeptide provides a mechanism for enhanced IL-1β in type 2
diabetes. Nat Immunol. 11:897–904. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chávez-Sánchez L, Garza-Reyes MG,
Espinosa-Luna JE, Chávez-Rueda K, Legorreta-Haquet MV and
Blanco-Favela F: The role of TLR2, TLR4 and CD36 in macrophage
activation and foam cell formation in response to oxLDL in humans.
Hum Immunol. 75:322–329. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Björkbacka H, Kunjathoor VV, Moore KJ, et
al: Reduced atherosclerosis in MyD88-null mice links elevated serum
cholesterol levels to activation of innate immunity signaling
pathways. Nat Med. 10:416–421. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
West XZ, Malinin NL, Merkulova AA, et al:
Oxidative stress induces angiogenesis by activating TLR2 with novel
endogenous ligands. Nature. 467:972–976. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Miller YI, Viriyakosol S, Binder CJ,
Feramisco JR, Kirkland TN and Witztum JL: Minimally modified LDL
binds to CD14, induces macrophage spreading via TLR4/MD-2, and
inhibits phagocytosis of apoptotic cells. J Biol Chem.
278:1561–1568. 2003. View Article : Google Scholar
|
|
25
|
Mullick AE, Tobias PS and Curtiss LK:
Modulation of atherosclerosis in mice by Toll-like receptor 2. J
Clin Invest. 115:3149–3156. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chávez-Sánchez L, Chávez-Rueda K,
Legorreta-Haquet MV, et al: The activation of CD14, TLR4, and TLR2
by mmLDL induces IL-1β, IL-6, and IL-10 secretion in human
monocytes and macrophages. Lipids Health Dis. 9:1172010. View Article : Google Scholar
|
|
27
|
Wiley JS, Sluyter R, Gu BJ, Stokes L and
Fuller SJ: The human P2X7 receptor and its role in innate immunity.
Tissue Antigens. 78:321–332. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Benetti E, Chiazza F, Patel NS and Collino
M: The NLRP3 Inflammasome as a novel player of the intercellular
crosstalk in metabolic disorders. Mediators Inflamm.
2013:6786272013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Solini A, Menini S, Rossi C, et al: The
purinergic 2X7 receptor participates in renal inflammation and
injury induced by high-fat diet: possible role of NLRP3
inflammasome activation. J Pathol. 231:342–353. 2013.PubMed/NCBI
|
|
30
|
Hussen J, Düvel A, Koy M and Schuberth HJ:
Inflammasome activation in bovine monocytes by extracellular ATP
does not require the purinergic receptor P2X7. Dev Comp Immunol.
38:312–320. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dekali S, Divetain A, Kortulewski T, et
al: Cell cooperation and role of the P2X7 receptor in pulmonary
inflammation induced by nanoparticles. Nanotoxicology. 7:1302–1314.
2013. View Article : Google Scholar
|
|
32
|
Usui F, Shirasuna K, Kimura H, et al:
Critical role of caspase-1 in vascular inflammation and development
of atherosclerosis in Western diet-fed apolipoprotein E-deficient
mice. Biochem Biophys Res Commun. 425:162–168. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Qiao Y, Wang P, Qi J, Zhang L and Gao C:
TLR-induced NF-κB activation regulates NLRP3 expression in murine
macrophages. FEBS Lett. 586:1022–1026. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Matsuura E, Lopez LR, Shoenfeld Y and Ames
PR: β2-glycoprotein I and oxidative inflammation in early
atherogenesis: a progression from innate to adaptive immunity?
Autoimmun Rev. 12:241–249. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lin J, Shou X, Mao X, et al: Oxidized low
density lipoprotein induced caspase-1 mediated pyroptotic cell
death in macrophages: implication in lesion instability? PLoS One.
8:e621482013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ehara S, Ueda M, Naruko T, et al: Elevated
levels of oxidized low density lipoprotein show a positive
relationship with the severity of acute coronary syndromes.
Circulation. 103:1955–1960. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lu B, Nakamura T, Inouye K, et al: Novel
role of PKR in inflammasome activation and HMGB1 release. Nature.
488:670–674. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Oury C: CD36: linking lipids to the NLRP3
inflammasome, atherogenesis and atherothrombosis. Cell Mol Immunol.
11:8–10. 2014. View Article : Google Scholar :
|
|
39
|
Sheedy FJ, Grebe A, Rayner KJ, et al: CD36
coordinates NLRP3 inflammasome activation by facilitating
intracellular nucleation of soluble ligands into particulate
ligands in sterile inflammation. Nat Immunol. 14:812–820. 2013.
View Article : Google Scholar : PubMed/NCBI
|