Introduction
Lung cancer is the leading cause of cancer death
worldwide. In fact, over 90% of deaths from solid tumors, including
lung cancer, is mainly due to cancer metastasis (1). Metastasis of tumor cells is
associated with epithelial-mesenchymal transition (EMT), which is a
process whereby epithelial cells acquire new features of mesenchyme
(2).
EMT is vital for the conversion of early-stage
tumors into invasive malignancies (3). Alterations in morphology, cellular
architecture, adhesion, and migration capacity are required for EMT
(4). As one of the hallmarks of
EMT, the functional loss of E-cadherin is currently thought to
promote invasion during carcinoma progression (5). The E-cadherin repressors, in
particular, Snail can be induced by a number of distinct signaling
pathways, such as TGF-β, Wnt and Notch (6–8).
TGF-β signaling is a primary inducer of EMT in
various cancers, including lung cancer (9–11).
TGF-β binding to its receptors leads to phosphorylation of Smad2
and Smad3, which partner with Smad4 and then translocate into the
nucleus where Smad transcriptional complexes control the expression
of target genes, including Snail (8,12).
Importantly, Smad3 and Smad4 rather than Smad2 played an essential
role in TGF-β-induced Snail expression and EMT (9,13–15).
Interleukin-6 (IL-6)-mediated aberrant activation of
Janus kinases/signal transducer and activator of transcription 3
(JAK/STAT3) signaling is frequently presented in human cancer
including lung cancer and implicated in transformation,
tumorigenicity, EMT and metastasis (16–20).
Additionally, inhibition of JAK2 tyrosine kinase or blockade of
activated Stat3 significantly suppressed the EMT process, cancer
cell migration and invasion in vitro, and attenuated cancer
cell metastasis in vivo (21–23).
Although no association of JAK/STAT3 signaling with EMT of lung
cancer is so far well established, elevated level of serum IL-6 has
been observed in lung cancer patients when compared with normal
donors and correlates with advanced lung cancer stage and an
overall poor prognosis (24,25),
suggesting that the IL-6/JAK/STAT3 pathway may participate in the
progression of lung cancer via EMT.
Recent studies have demonstrated that TGF-β-mediated
cancer metastasis initiation was associated with the activation of
JAK/STAT3 pathway in colorectal cancer (26). Taken together, we hypothesized that
IL-6/JAK/STAT3 signaling is required for TGF-β-mediated EMT in lung
cancer.
Materials and methods
Cell culture
Human lung cell lines A549 and H1650 were purchased
from Cell Bank of Chinese Academy of Science. Cells were cultured
in Roswell Park Memorial Institute (RPMI)-1640 medium (Hyclone)
with 50 U/ml each of penicillin and streptomycin and 10%
heat-inactivated fetal bovine serum (FBS, Invitrogen) in humidified
incubators at the condition of 37°C and 5% CO2.
Reagents and antibodies
Human recombinant TGF-β1 and IL-6 were obtained from
R&D Systems Inc. (Minneapolis, MN, USA). TGF-β1 was diluted in
sterile 4 mM HCl containing 1 mg/ml human serum albumin (HSA); IL-6
was reconstituted in sterile PBS containing 0.1% HSA. JAK2
inhibitor AG490 and Smad3 phosphorylation inhibitor SIS3 were
purchased from Merck KGaA (Darmstadt, Germany) and Santa Cruz
Biotechnology (Santa Cruz, CA, USA), respectively. Antibodies used
for western blotting were mouse anti-E-cadherin (1:3,000, BD
Biosciences), mouse anti-vimentin (1:4,000, BD Biosciences), rabbit
anti-Snail (1:1,000, Cell Signaling), rabbit anti-Smad3 (1:1,000,
Cell Signaling), rabbit anti-phospho-Smad3 (Ser423/425, 1:1,000,
Cell Signaling), rabbit anti-Stat3 (1:1,000, Cell Signaling),
rabbit anti-phospho-Stat3 (Tyr705, 1:1,000, Cell Signaling), mouse
anti-GAPDH (1:3,000, Abcam).
Western blot analysis
Western blot analysis was performed according to the
protocol described by us (27)
with some modifications. Briefly, cells were lysed in a RIPA buffer
with protease inhibitor cocktail (Sigma-Aldrich) and phosphatase
inhibitor cocktail (Sigma-Aldrich), and then clarified by
centrifugation. Total cell lysates were resuspended in SDS sample
buffer and resolved by SDS-PAGE. Proteins were transferred to
nitrocellulose membrane (Millipore, Bedford, MA, USA) and then
blocked with 5% BSA/TBST buffer for 1 h at room temperature.
Membranes were incubated with primary antibodies overnight before
incubation with the corresponding HRP-conjugated secondary
antibodies and detected using the SuperSignal West Pico
Chemiluminescent Substrate (Thermo Fisher Scientific Inc.). All
experiments were performed in triplicate.
RNA extraction and quantitative real-time
PCR (qRT-PCR)
Cells were grown to 80% confluence on 6-well plates
and subjected to RNA extraction using 1.0 ml RNAiso Plus (Takara)
according to the manufacturer’s instructions. Synthesis of cDNA
with reverse transcriptase was performed by M-MLV First Strand kit
(Invitrogen). cDNA aliquots were subjected to qRT-PCR reactions
using the Platinum®SYBR®Green
qPCRsupermix-UDG with ROX (Invitrogen) on ABI PRISM 7500 Sequence
Detection System (Applied Biosystems). Primers used for qRT-PCR
were as follows: 5′-CGAAAGGCCTTCA ACTGCAAAT-3′ (forward) and
5′-ACTGGTACTTCTTGA CATCTG-3′ (reverse) for Snail; and
5′-TGCACCACCAACTG CTTAGC-3′ (forward) and 5′-GGCATGGACTGTGGTCAT
GAG-3′ (reverse) for GAPDH. The PCR program was 50°C for 2 min,
95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C
for 1 min. A standard melting-curve analysis was carried out at the
end of the amplification. Each qRT-PCR experiment was done in
triplicates. All the mRNA expression values were normalized to an
internal control GAPDH.
Luciferase reporter gene construct
The promoter region of PAI-1, −960 to +124, was
amplified by PCR with primers (forward: CGGGGTACCGCACACCCTGCAAACCTGCC and
reverse: CCGCTCGAGCGATTGGCGGTTCGTCCTG,
containing a KpnI site and a XhoI site,
respectively). After digestion with KpnI and XhoI,
the resultant fragments were directly ligated into the pGL3-basic
luciferase vector (Promega). Before transfection, the sequence of
plasmid construct was confirmed by direct sequencing.
Transient transfection and reporter gene
assays
Cells were co-transfected with 800 ng pgl3-basic
constructs with PAI-1 promoter and 16 ng SV-40 plasmid (as a
normalizing control) using Lipofectamine 2000 (Invitrogen). Four
hours later, the cells were treated with or without 50 μM
AG490 and then incubated for 18 h in the presence or absence of 5
ng/ml TGF-β1. Finally, luciferase activity of the transfected cells
was determined using the Dual-Luciferase Reporter Assay System
(Promega) on a TD20/20 Luminometer (Turner Designs, Sunnyvale, CA,
USA). Three independent transfection experiments were carried out,
and each was done in triplicate. Results are reported as relative
luciferase activities, which are obtained by dividing firefly
luciferase activity with SV-40 luciferase activity.
Cell invasion and migration assays
For invasion assay, BioCoat Matrigel invasion
chambers (BD Biosciences) with 8-μm pore polycarbonate
membranes were pre-treated with serum-free RPMI-1640
(Gibco-Invitrogen) medium at 37°C for two hours. After removing the
medium, we added 750-μl medium with 10% FBS as
chemoattractant to each lower chamber, and then added
5×104 cells with 1% FBS medium to each upper chamber.
Two hours later, the regents (5 ng/ml TGF-β1, 50 μM AG490 or
50 ng/ml IL-6) were added to the upper chambers, and further
incubated at 37°C for 12 h. The inserts were removed and
non-invasive cells on the upper surface were removed by a cotton
swab. The invasive cells on the lower surface of the membrane were
then fixed in 100% methanol for 15 min, air-dried, and stained with
1% crystal violet. Cells from three microscopic fields were
photographed and counted. For migration assay, similar procedure to
the invasion assay was conducted but omitting addition of Matrigel.
Each experiment was done in triplicate.
Statistical analysis
Data are presented as the mean ± standard deviations
(SD) and Student’s t-test was performed to determine statistically
significant differences between two groups. Statistical differences
were considered to be significant at P<0.05. All the statistical
analysis was performed using GraphPad Prism 5.01 software (GraphPad
Software, Inc.) and STATA 10.1 software (Stata Corp, College
Station).
Results
Snail is involved in TGF-β-induced
EMT
The A549 cell line has been widely used as a model
system to study the mechanisms of carcinogenesis and tumor
progression in lung cancer. First, we observed the morphological
changes in A549 cells in the presence of TGF-β1. In the absence of
TGF-β1, A549 cells maintained a classic epithelial morphology;
however, after treatment with TGF-β1 for 24 h, A549 cells displayed
a spindle-shape, fibroblast-like morphology (Fig. 1A). Next, we used western blot assay
to investigate the expression of epithelial marker, E-cadherin, and
the mesenchymal marker vimentin. The results showed that the
expression of E-cadherin was significantly decreased and the
expression of vimentin was increased with TGF-β1 treatment for 24 h
in A549 cells (Fig. 1B).
 | Figure 1.TGF-β-induced EMT is dependent on
p-Smad3 and Snail in human lung cancer cells A549. Cells were
serum-starved for 24 h before treatments. (A) TGF-β1 exposure
induced transition of the epithelial to the mesenchymal-like
phenotype in A549 cells. Cells were treated with 1% FBS without
TGF-β1 (upper), 1% FBS with 2 ng/ml TGF-β1 (middle), and 1% FBS
with 5 ng/ml TGF-β1 (lower). Cell morphology was examined at 24 h
after TGF-β1 treatment and photographed using a phase-contrast
microscope. (B) TGF-β1 altered expression of EMT molecular markers.
Cells were treated with or without TGF-β1 for 24 h; total cell
lysates were extracted and subjected to western blot analyses for
expression of E-cadherin, vimentin and Snail. GAPDH was used as an
internal control. (C) TGF-β1 upregulated mRNA expression of Snail.
Cells were exposed to 5 ng/ml TGF-β1 for the indicated times, and
mRNA level of Snail was determined by quantitative RT-PCR. (D)
TGF-β1 phosphorylated Smad3 and upregulated protein level of Snail
expression. Cells were treated with or without 5 ng/ml TGF-β1 for
the indicated times, and p-Smad3, Smad3 and Snail protein
expressions were detected by western blotting. (E) SIS3 inhibited
TGF-β-induced phosphorylation of Smad3 and restored expression of
EMT molecular markers in the presence of TGF-β. Cells were
pretreated with 3 μM SIS3 (Smad3 phosphorylation inhibitor)
for 4 h, and then exposed to 5 ng/ml TGF-β1 for 24 h, the levels of
E-cadherin, vimentin, p-Smad3, Smad3 and Snail were determined by
western blotting. |
Snail plays a vital role in regulating EMT during
tumor progression (7,8). Therefore, we investigated whether
expression of Snail was involved in TGF-β-induced EMT in A549
cells. As a result, TGF-β1 treatment increased Snail mRNA
expression, which showed the highest level at 1 h (Fig. 1C). The inducing effects of TGF-β on
Snail were also seen at protein level in A549 cells (Fig. 1B and D). Taken together, these data
indicated that TGF-β was able to upregulate the expression of Snail
and subsequently induce EMT in lung cancer cells.
Phosphorylated Smad3 is required for
TGF-β-induced EMT
Although Smad3 is the key to TGF-β-induced EMT
(13,15), the role of phosphorylated Smad3
(p-Smad3) in TGF-β-induced EMT has not been highlighted. To address
this, we inhibited the activation of Smad3 by a specific inhibitor
SIS3 (28) and found that SIS3
could remarkably decrease TGF-β-induced phosphorylation of Smad3
(Fig. 1E). SIS3 significantly
restored E-cadherin expression and impaired vimentin and Snail
expression in the presence of TGF-β1 (Fig. 1E). These results suggested that
TGF-β-induced EMT depends on p-Smad3 and Snail in lung cancer
cells.
JAK/STAT3 signaling is required for TGF-β-mediated
transcriptional responses and EMT. Based on the notion that
TGF-β-induced metastasis initiation requires the participation of
JAK/STAT3 signaling pathway in colorectal cancer (26), we hypothesize that the JAK/STAT3
signaling is necessary for TGF-β-mediated EMT. To test this
hypothesis, we first used a JAK2-specific inhibitor AG490 to
expectably suppress the JAK/STAT3 signaling activity and found that
AG490 can significantly depress the phosphorylation of Stat3
(p-Stat3) in lung cancer cell lines A549 and H1650 (Fig. 2A). Secondly and importantly, AG490
markedly reduced Smad3 phosphorylation induced by TGF-β1 (Fig. 2B). Thirdly, we performed luciferase
reporter assay to assess whether JAK/STAT3 pathway could regulate
TGF-β-induced Smad transcriptional activity in A549 and H1650
cells. Cells transfected with Smad-mediated PAI-1 reporter plasmid
were stimulated with TGF-β in the presence and absence of AG490. As
illustrated in Fig. 2C, TGF-β1 can
significantly increase the luciferase reporter activity, and AG490
treatment significantly attenuated both the basal and TGF-β-induced
PAI-1 promoter activation. Fourthly, AG490 can diminish
TGF-β-induced increase in Snail and MMP2 (Fig. 2B, D and E), which have been widely
recognized to promote cancer metastasis. These data demonstrate
that JAK/STAT3 signaling is required for TGF-β-induced
phosphorylation of Smad3, Smad-mediated transcriptional responses
and EMT in lung cancer.
AG490 blocks TGF-β-induced cell migration
and invasion
Given the facts that TGF-β signaling can stimulate
the migration and invasion of tumor cells (29), and the JAK/STAT3 signaling is
implicated in the regulation of cell migration and invasion
(30), we adopted transwell assays
to examine whether JAK/STAT3 signaling is involved in TGF-β-induced
migration and invasion in A549 and H1650 cells. As a result, TGF-β1
enhanced the migratory ability of the two cell lines, and AG490
blocked both basal and TGF-β-stimulated cell migration (Fig. 3A). Moreover, AG490 also blocked
tumor cell invasion induced by TGF-β1 (Fig. 3B). These results suggested that the
basal JAK/STAT3 signaling was required for TGF-β-induced migration
and invasion of lung cancer cells.
IL-6 enhances TGF-β-induced EMT in lung
cancer cells
According to our findings described above, we
speculated that enlargement of the JAK/STAT3 signaling might
enhance the TGF-β/Smad signaling and TGF-β-induced EMT. Therefore,
we used human recombinant IL-6 to enhance the JAK/STAT3 signaling
pathway (Fig. 4A), and found that
IL-6 stimulation increased phosphorylation levels of Smad3 in cells
treated with TGF-β1, albeit total Smad3 and Smad4 levels did not
alter in cells treated with IL-6 and/or TGF-β1 (Fig. 4B). Next, we examined the effect of
IL-6 and/or TGF-β1 treatment on the expression of Snail, the
important EMT inducer, in A549 and H1650 cells. After serum
starvation for 24 h, Snail mRNA levels were rapidly induced upon
TGF-β1 treatment in A549 and H1650 cells, but not in IL-6 treated
cells (Fig. 4C). More importantly,
Snail expression was higher in cells treated with TGF-β1 and IL-6
than TGF-β1 alone (Fig. 4C and
D).
Subsequently, we evaluated the synergistic effect of
IL-6 and TGF-β on the induction of EMT in lung cancer cells and
found that the expression of E-cadherin was almost abrogated in
cells treated with TGF-β1 and IL-6, and the expression of vimentin
was higher in cells treated with TGF-β1 and IL-6 than TGF-β1 alone
(Fig. 4E). Taken together, these
data showed that the IL-6/JAK/STAT3 signaling enhanced TGF-β/Smad
pathway and then accelerated the TGF-β-induced EMT process in lung
cancer cells.
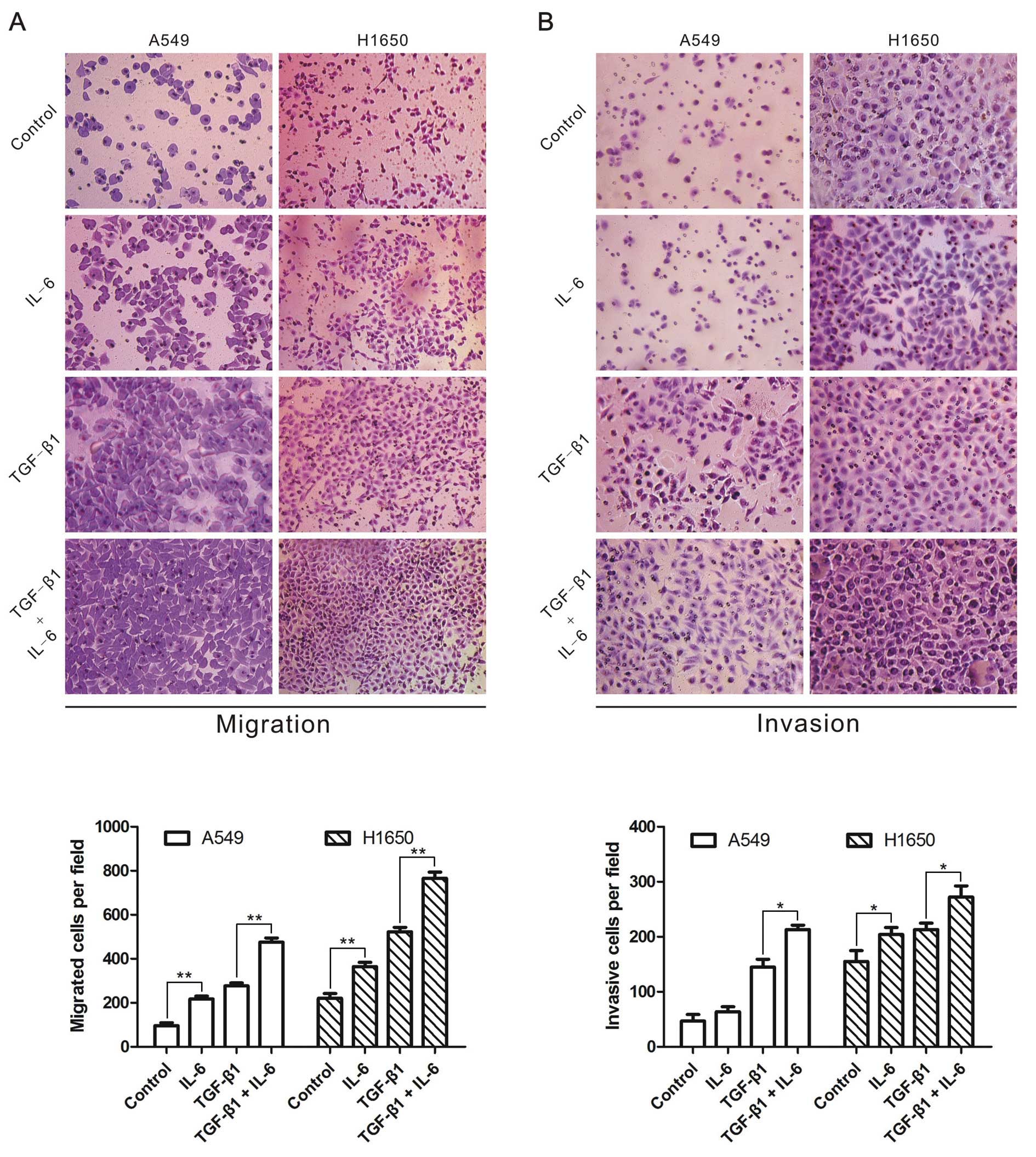
IL-6 increases TGF-β-induced cell
migration and invasion
Finally, we determined whether IL-6 strengthened
TGF-β-induced migration and invasion in A549 and H1650 cells
treated with TGF-β1 and/or IL-6, and observed that IL-6 or TGF-β1
enhanced the migration and invasion of lung cancer cells (Fig. 5). More importantly, cells treated
with TGF-β1 in combination with IL-6 showed higher capability of
migration and invasion than TGF-β1 alone (Fig. 5). The results indicated that IL-6
was able to enhance TGF-β-induced lung cancer cell migration and
invasion.
Discussion
EMT is a critical event occurring during cancer
metastasis (3). Increasing number
of studies have yielded distinct signaling pathways that regulate
EMT (31). In this study, we
provided new insight into the contribution of an interaction
between the JAK/STAT3 and TGF-β/Smad pathways to the induction of
EMT in lung cancer. To the best of our knowledge, this is the first
evidence of identifying that the JAK/STAT3 pathway can enhance
TGF-β-induced EMT.
It has been demonstrated that the JAK/STAT3 pathway
is required to sustain EGF/EGFR-induced EMT-associated phenotypes
in ovarian and breast cancers (32,33).
Moreover, Yamashita et al established a molecular link
between Stat3, LIV1, and Snail during the EMT of gastrula organizer
cells in zebrafish embryos (34).
Our results support the potential role of the JAK/STAT3 signaling
in promoting EMT, which may be vital for cancer invasion and
metastasis.
In the process of EMT, epithelial cells acquire
expression of mesenchymal components and manifest migratory and
invasive phenotypes (35). As a
functional consequence of TGF-β signaling, decreased expression of
E-cadherin, increased expression of vimentin and increased
abilities of migration and invasion were displayed in lung cancer
cells A549 and H1650 (Figs. 4 and
5). These findings are in
accordance with previous studies that TGF-β can induce EMT in
certain types of cancer cells (9,36–38).
Specifically, we found that TGF-β can increase the expression of
Snail at both mRNA and protein levels in lung cancer cells,
consisting with the findings obtained in pancreatic cancer Panc-1
cells (39), Madin-Darby canine
kidney cells (40), retinal
pigment epithelia (41) and mouse
mammary epithelial HMuMg cells (42). Therefore, upregulation of Snail can
be a common mechanism underlying TGF-β-induced EMT.
Besides TGF-β, IL-6 was reported to induce EMT in
breast, colorectal and prostate cancer cells (16–18).
In lung cancer cells, we did not find that IL-6 directly elicited
EMT. This discrepancy may be explained by the following three
aspects: first, here we used low dose of IL-6 to treat cells,
unlike Sullivan et al who ectopically expressed IL-6 to
enlarge its secretion (16).
Second, this difference depends on the context of certain types of
cancer cells. Third, IL-6 might be indirectly involved in the EMT
process in lung cancer cells. In support of this, we found that
IL-6 promoted the EMT process induced by TGF-β, suggesting the
cooperation of IL-6/JAK/STAT3 with TGF-β/Smad signaling pathways in
EMT.
Our results showed that IL-6 boosted the expression
of Snail induced by TGF-β/Smad pathway, contributing greatly to EMT
(8,43). It is worth to notice that
TGF-β-induced Snail transcription was highly dependent on the
cooperation of p-Smad with c-Myc, which was required for rapid
Snail activation upon Smad (14).
Significantly, IL-6 is capable of inducing c-Myc expression through
activating Stat3 which binds directly to the promoter of c-Myc
(44,45). Therefore, IL-6 can enhance
TGF-β-induced Snail both c-Myc- and Smad-dependently.
Notably, in EGFR-overexpressing tumor cells,
activated Stat3 is responsible for EGF-mediated desensitization of
the TGF-β signaling (46). In
contrast, we focused on A549 cells expressing lower EGFR and H1650
cells harboring mutated EGFR, and found that the IL-6-induced Stat3
activation enhanced the TGF-β/Smad signaling. Similarly, IL-6
regulation of TGF-β receptor compartmentalization and turnover
enhances TGF-β1 signaling in human renal epithelial cells (47). These findings suggest that
different effects of IL-6 and/or Stat3 on TGF-β signaling depends
upon context of cell types, further investigations are warranted to
uncover the underlying mechanisms.
In conclusion, our findings identify that the
JAK/STAT3 pathway is required for TGF-β-induced EMT and lung cancer
cell migration and invasion, and the IL-6/JAK/STAT3 and TGF-β/Smad
signaling can synergistically enhance EMT in lung carcinomas. This
study suggests a new rationale for inhibiting cancer metastasis
using anti-IL-6/JAK/STAT3 and anti-TGF-β/Smad therapeutic
strategies.
Acknowledgements
This study was supported in part by
the grants from National Natural Science Foundation of China
(81372277 and 81171894 to H.-T. Zhang; 81201575 to Z. Liu), Program
for New Century Excellent Talents in University (NCET-09-0165 to
H.-T. Zhang), the Jiangsu Province Key Provincial Talents Program
(RC2011106 to J. Zhao), ‘333’ Project of Jiangsu Province
Government (to H.-T. Zhang), Soochow Scholar Project of Soochow
University (to H.-T. Zhang), and Suzhou Key Laboratory for
Molecular Cancer Genetics (SZS201209 to H.-T. Zhang).
References
|
1.
|
Gupta GP and Massague J: Cancer
metastasis: building a framework. Cell. 127:679–695. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Kang Y and Massague J:
Epithelial-mesenchymal transitions: twist in development and
metastasis. Cell. 118:277–279. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Lee JM, Dedhar S, Kalluri R and Thompson
EW: The epithelialmesenchymal transition: new insights in
signaling, development, and disease. J Cell Biol. 172:973–981.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelialmesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Peinado H, Olmeda D and Cano A: Snail, Zeb
and bHLH factors in tumour progression: an alliance against the
epithelial phenotype? Nat Rev Cancer. 7:415–428. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Vincent T, Neve EP, Johnson JR, et al: A
SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta
mediated epithelial-mesenchymal transition. Nat Cell Biol.
11:943–950. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Horiguchi K, Sakamoto K, Koinuma D, et al:
TGF-beta drives epithelial-mesenchymal transition through
deltaEF1-mediated downregulation of ESRP. Oncogene. 31:3190–3201.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Pirozzi G, Tirino V, Camerlingo R, et al:
Epithelial to mesenchymal transition by TGFbeta-1 induction
increases stemness characteristics in primary non small cell lung
cancer cell line. PLoS One. 6:e215482011. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Massague J and Chen YG: Controlling
TGF-beta signaling. Genes Dev. 14:627–644. 2000.
|
|
13.
|
Roberts AB, Tian F, Byfield SD, et al:
Smad3 is key to TGF-beta-mediated epithelial-to-mesenchymal
transition, fibrosis, tumor suppression and metastasis. Cytokine
Growth Factor Rev. 17:19–27. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Smith AP, Verrecchia A, Faga G, et al: A
positive role for Myc in TGFbeta-induced Snail transcription and
epithelial-to-mesenchymal transition. Oncogene. 28:422–430. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Reka AK, Kurapati H, Narala VR, et al:
Peroxisome proliferator-activated receptor-gamma activation
inhibits tumor metastasis by antagonizing Smad3-mediated
epithelial-mesenchymal transition. Mol Cancer Ther. 9:3221–3232.
2010. View Article : Google Scholar
|
|
16.
|
Sullivan NJ, Sasser AK, Axel AE, et al:
Interleukin-6 induces an epithelial-mesenchymal transition
phenotype in human breast cancer cells. Oncogene. 28:2940–2947.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Rojas A, Liu G, Coleman I, et al: IL-6
promotes prostate tumori-genesis and progression through autocrine
cross-activation of IGF-IR. Oncogene. 30:2345–2355. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Xiong H, Hong J, Du W, et al: Roles of
STAT3 and ZEB1 proteins in E-cadherin down-regulation and human
colorectal cancer epithelial-mesenchymal transition. J Biol Chem.
287:5819–5832. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Bromberg J and Wang TC: Inflammation and
cancer: IL-6 and STAT3 complete the link. Cancer Cell. 15:79–80.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Looyenga BD, Hutchings D, Cherni I,
Kingsley C, Weiss GJ and Mackeigan JP: STAT3 is activated by JAK2
independent of key oncogenic driver mutations in non-small cell
lung carcinoma. PLoS One. 7:e308202012. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Huang C, Yang G, Jiang T, Zhu G, Li H and
Qiu Z: The effects and mechanisms of blockage of STAT3 signaling
pathway on IL-6 inducing EMT in human pancreatic cancer cells in
vitro. Neoplasma. 58:396–405. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Xie TX, Huang FJ, Aldape KD, et al:
Activation of stat3 in human melanoma promotes brain metastasis.
Cancer Res. 66:3188–3196. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Abdulghani J, Gu L, Dagvadorj A, et al:
Stat3 promotes meta-static progression of prostate cancer. Am J
Pathol. 172:1717–1728. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Enewold L, Mechanic LE, Bowman ED, et al:
Serum concentrations of cytokines and lung cancer survival in
African Americans and Caucasians. Cancer Epidemiol Biomarkers Prev.
18:215–222. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Pine SR, Mechanic LE, Enewold L, et al:
Increased levels of circulating interleukin 6, interleukin 8,
C-reactive protein, and risk of lung cancer. J Natl Cancer Inst.
103:1112–1122. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Calon A, Espinet E, Palomo-Ponce S, et al:
Dependency of colorectal cancer on a TGF-beta-driven program in
stromal cells for metastasis initiation. Cancer Cell. 22:571–584.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Liu XL, Xiao K, Xue B, et al: Dual role of
TGFBR3 in bladder cancer. Oncol Rep. 30:1301–1308. 2013.PubMed/NCBI
|
|
28.
|
Jinnin M, Ihn H and Tamaki K:
Characterization of SIS3, a novel specific inhibitor of Smad3, and
its effect on transforming growth factor-beta1-induced
extracellular matrix expression. Mol Pharmacol. 69:597–607. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Massague J: TGFbeta in cancer. Cell.
134:215–230. 2008. View Article : Google Scholar
|
|
30.
|
Sansone P and Bromberg J: Targeting the
interleukin-6/Jak/stat pathway in human malignancies. J Clin Oncol.
30:1005–1014. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013.PubMed/NCBI
|
|
32.
|
Lo HW, Hsu SC, Xia W, et al: Epidermal
growth factor receptor cooperates with signal transducer and
activator of transcription 3 to induce epithelial-mesenchymal
transition in cancer cells via up-regulation of TWIST gene
expression. Cancer Res. 67:9066–9076. 2007. View Article : Google Scholar
|
|
33.
|
Colomiere M, Ward AC, Riley C, et al:
Cross talk of signals between EGFR and IL-6R through JAK2/STAT3
mediate epithelial-mesenchymal transition in ovarian carcinomas. Br
J Cancer. 100:134–144. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Yamashita S, Miyagi C, Fukada T, Kagara N,
Che YS and Hirano T: Zinc transporter LIVI controls
epithelial-mesenchymal transition in zebrafish gastrula organizer.
Nature. 429:298–302. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: at the crossroads of development
and tumor metastasis. Dev Cell. 14:818–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Prunier C and Howe PH: Disabled-2 (Dab2)
is required for transforming growth factor beta-induced epithelial
to mesenchymal transition (EMT). J Biol Chem. 280:17540–17548.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Cho HJ, Baek KE, Saika S, Jeong MJ and Yoo
J: Snail is required for transforming growth factor-beta-induced
epithelial-mesenchymal transition by activating PI3 kinase/Akt
signal pathway. Biochem Biophys Res Commun. 353:337–343. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Shintani Y, Maeda M, Chaika N, Johnson KR
and Wheelock MJ: Collagen I promotes epithelial-to-mesenchymal
transition in lung cancer cells via transforming growth factor-beta
signaling. Am J Respir Cell Mol Biol. 38:95–104. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Horiguchi K, Shirakihara T, Nakano A,
Imamura T, Miyazono K and Saitoh M: Role of Ras signaling in the
induction of snail by transforming growth factor-beta. J Biol Chem.
284:245–253. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Peinado H, Quintanilla M and Cano A:
Transforming growth factor beta-1 induces snail transcription
factor in epithelial cell lines: mechanisms for epithelial
mesenchymal transitions. J Biol Chem. 278:21113–21123. 2003.
View Article : Google Scholar
|
|
41.
|
Li H, Wang H, Wang F, Gu Q and Xu X: Snail
involves in the transforming growth factor beta1-mediated
epithelial-mesenchymal transition of retinal pigment epithelial
cells. PLoS One. 6:e233222011. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Thuault S, Tan EJ, Peinado H, Cano A,
Heldin CH and Moustakas A: HMGA2 and Smads co-regulate SNAIL1
expression during induction of epithelial-to-mesenchymal
transition. J Biol Chem. 283:33437–33446. 2008. View Article : Google Scholar
|
|
43.
|
Shih JY and Yang PC: The EMT regulator
slug and lung carcino-genesis. Carcinogenesis. 32:1299–1304. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Dauer DJ, Ferraro B, Song L, et al: Stat3
regulates genes common to both wound healing and cancer. Oncogene.
24:3397–3408. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Kiuchi N, Nakajima K, Ichiba M, et al:
STAT3 is required for the gp130-mediated full activation of the
c-myc gene. J Exp Med. 189:63–73. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Luwor RB, Baradaran B, Taylor LE, et al:
Targeting Stat3 and Smad7 to restore TGF-beta cytostatic regulation
of tumor cells in vitro and in vivo. Oncogene. 32:2433–2441. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Zhang XL, Topley N, Ito T and Phillips A:
Interleukin-6 regulation of transforming growth factor (TGF)-beta
receptor compartmentalization and turnover enhances TGF-beta1
signaling. J Biol Chem. 280:12239–12245. 2005. View Article : Google Scholar : PubMed/NCBI
|