LncRNA microarray profiling identifies novel circulating lncRNAs in hidradenitis suppurativa
- Authors:
- Published online on: May 2, 2024 https://doi.org/10.3892/mmr.2024.13236
- Article Number: 112
-
Copyright: © De Felice et al. This is an open access article distributed under the terms of Creative Commons Attribution License.
Abstract
Introduction
Long noncoding RNAs (lncRNAs) are a class of non-protein-coding RNAs >200 nucleotides in size. lncRNAs can be classified into two groups: Small ncRNAs, such as microRNAs (miRNAs), small interfering RNAs, small nucleolar RNAs, Piwi-interacting RNAs, small nuclear RNAs and tRNA-derived fragments, and lncRNAs (1). Compared with miRNAs, which are responsible for the downregulation or upregulation of genes, lncRNAs, despite being poorly conserved, repress or activate gene expression, thereby fulfilling an important role in the complexity of species change (1,2). LncRNAs are involved in genome stability, the formation of cellular organelles, structural functions and gene expression regulation via interacting with nucleic acids and/or proteins (1). The majority of lncRNAs are localized in the nucleus, and their function at the epigenetic level can be accounted for in different ways. Signal lncRNAs, under various stimuli, recruit histone methyltransferases and induce chromatin methylation and transcriptional silencing (2,3). Furthermore, decoy lncRNAs are responsible for gene repression, as they limit the availability of splicing factors (2,3). In addition, guide lncRNAs select specific ribonucleoproteins that regulate specific target sites and functions, whereas scaffold lncRNAs bind different nuclear components, and, according to their function, either promote or repress transcription (2–4). Finally, enhancer lncRNAs interact with enhancer or promoter regions, leading to the modulation of gene expression through the formation of chromosomal loops (5). Previous studies have also reported that lncRNAs are involved in a wide range of biological activities, including transcriptional and post-transcriptional processes, and epigenetics (6). Furthermore, they have been shown to be involved in physiological and/or pathological biological processes (6). LncRNAs have been demonstrated to have important roles in various types of cancer, including bladder cancer (6), ovarian cancer (7), pancreatic cancer (8), breast cancer and metastasis (9). LncRNAs also fulfill roles in various degenerative conditions, including Alzheimer's disease (10), osteoarthritis (11), multiple sclerosis (12), but also in autoimmunity states, such as Sjogren's syndrome (13), autoimmune hepatitis (14) and types 1 and 2 diabetes (15). An increasing amount of data are showing that lncRNAs exert a critical role in the biology of skin and in the pathology of cutaneous diseases (16).
Hidradenitis suppurativa (HS), also termed acne inversa, is an inflammatory skin condition that affects ~1% of the worldwide population, and its onset occurs at around the age of 20 years in patients (16). Western women have a prevalence of HS of 3:1 with respect to men, whereas in Asia, the ratio is inverted 1:2 in favor of men (17). Commonly affected regions are the buttocks in men, and the axillae, groins and breasts in women (17). The manifestation of HS passes through three stages: The Hyperkeratosis of hair follicles with subsequent occlusion and dilation, epithelial wall rupture and infiltration of inflammatory cells, and subsequently, formation of interconnecting sinus tracts (17). Clinically, patients present with painful nodules, suppurative abscesses and comedones. Frequently, due to the presence of nodules and rupture of abscesses, patients experience a burning sensation, pain and malodor, with the discharge of purulent material; moreover, infections supported by bacteria induce HS chronicity (17,18). HS induces poor health, an impaired quality of life and a high risk of mortality in patients, as it is associated with several comorbidities. For this reason, screening is recommended if certain conditions or clinico-pathological characteristics are met in the patients, such as acne, pyoderma gangrenosum, smoking, polycystic ovary syndrome, cardiovascular disease, metabolic diseases, inflammatory bowel disease and spondyloarthritis (19).
Of note, obesity represents a primary risk factor for hidradenitis, although the exact of the association between the two disorders is not yet well understood. The chronic inflammatory state established in patients with obesity likely plays a role in contributing to the susceptibility to hidradenitis as obesity generates an imbalance in a plethora of cytokines and adipokines, contributing to dysregulation in the immune system, thus participating in the etiopathogenesis of hidradenitis (20,21).
In the present study, plasma samples from 25 patients with HS and matched healthy controls were collected and screened using genome-wide profiling microarray, and subsequently reverse transcription-quantitative (RT-q)PCR analysis was performed to investigate the presence of, and potentially increase the knowledge of, novel circulating lncRNA biomarkers for HS. Subsequently, lncRNA target predictions were made to associate the lncRNAs with their associated target mRNAs.
Patients and methods
Recruitment of patients
To profile circulating lncRNAs, 25 patients (19 females and 6 males) aged 27.07±14.76 years, and 25 healthy volunteers (18 females and 7 males) aged 33.25±11.26 years, were recruited from the Dermatology Unit of the University of Campania Luigi Vanvitelli (Caserta, Italy), and venous blood samples were collected from each participant in the study. Dysregulated lncRNAs in patients with HS were identified and used for further analysis and validation. The association between the selected lncRNAs and HS characteristics and prognosis was then investigated in the circulation of an expanded cohort of patients with HS (n=25) and healthy controls (n=25). Written informed consent was obtained from all the participants. The entire trial was conducted in agreement with the guidelines of The Ethics Committee of the University of Campania Luigi Vanvitelli, who also approved the research (approval no. 12478/20). The blood samples were handled in accordance with the guidelines of the Declaration of Helsinki.
Blood collection
A venous blood sample (10 ml) was collected and analyzed from each participant for the purposes of lncRNA identification and quantification. For human whole blood isolation, Lymphoprep™ (Axis-Shield Diagnostics, Ltd.) was used in accordance with the manufacturer's instructions.
RNA extraction
From whole blood samples, total RNAs were extracted using Invitrogen® TRIzol™ reagent (cat. no. 15596-026; Thermo Fisher Scientific, Inc.), according to the manufacturer's instructions. RNA isolated with this protocol was extracted from all white cells (polymorphonuclear leukocytes and mononuclear cells) and subjected to a DNase digestion step. A concentration of 1 µl DNase I (2 U; RNase-free, cat. no. AM2222-24; Thermo Fisher Scientific, Inc.) for up to 10 µg RNA was used, in line with the manufacturer's instructions. The RNA integrity was subsequently evaluated using standard denaturing 1% agarose gel electrophoresis and the SYBR green dye (Qiagen, Inc.).
LncRNA microarray profiling
For lncRNA profiling, human RT2 lncRNA PCR assays (Qiagen, Inc.) were applied. The RT2 First Strand Kit (Qiagen, Inc.) was used for RT, which included an exclusive genomic DNA elimination step to remove any remaining contamination. Prior to hybridization to the chip (Human lncFinder RT2 lncRNA PCR Array; cat. no. 330721; Qiagen Inc.) RT2 SYBR Green Mastermix for RT-qPCR (Qiagen, Inc.) was used. Microarrays were scanned using an Agilent microarray scanner (Agilent Technologies, Inc.), directed by GenePix Pro 6.0 software (Axon Instruments; Molecular Devices, LLC). Scanned images (in the .tiff format) were imported into Agilent Feature Extraction software v.12.2 (Agilent Technologies, Inc.) for grid alignment and expression data analysis. Expression data were corrected and normalized (according to the quartiles) using the robust multiarray average algorithm included in the Agilent software. Differentially expressed lncRNAs were then identified using fold change as the filter. The microarray data were selected applying threshold values of >2 and <−2-fold change under false discovery rate protection (P<0.05), and >25,000 human long noncoding transcripts were analyzed.
Validation of the gene expression of lncRNAs and lncRNAs co-expressed with mRNAs in the blood, as determined by RT-qPCR
RT-qPCR quantification of the expression of lncRNAs and lncRNAs co-expressed with mRNAs was performed using the PowerTrack SYBR Green Master Mix (Qiagen, Inc.) using an Applied Biosystems™ QuantStudio 5 instrument (Thermo Fisher Scientific, Inc.), following precisely the manufacturer's protocol with the following thermocycling conditions: Enzyme activation at 95°C for 2 min 1X, denaturation phase at 95°C for 14 sec, followed by annealing/extension at 60°C for 60 sec; the final two phases were repeated 40 times. Reactions were performed in a mixture (10 µl) containing 1 µl cDNA template, 5 µl 2X PowerTrack SYBR Green Master Mix and 0.5 µM each of sense and antisense primers. Table I shows the sequences of primers used for the RT-qPCR analysis. RT-qPCR reactions were performed in triplicate for each sample, and the specificity of the PCR products was estimated using the dissociation curve. β2-microglobulin was used as the internal control. For quantitative results, expression of each lncRNA was represented as the fold change using the 2−ΔΔCq method, and then analyzed for statistical significance (22,23).
Bioinformatics analysis
Through searching three databases, RNAinter (http://www.rnainter.org/), LncRRIsearch (http://rtools.cbrc.jp/LncRRIsearch/) and NPinter (http://bigdata.ibp.ac.cn/npinter4), potential mRNA targets for each lncRNA were identified with adjusted P-value was set to <0.05. To determine the roles of differentially expressed mRNAs, Gene Ontology (GO) analysis was applied to categorize mRNAs according to their involvement in biological processes, cellular components or molecular function. Subsequently, differentially regulated mRNAs were uploaded into the Database for Annotation, Visualization and Integrated Discovery (https://david.ncifcrf.gov) for annotation and functional analysis, including gene set enrichment analysis and mapping gene sets to the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway. The KEGG database (http://www.genome.ad.jp/kegg) and BioCarta (https://www.hsls.pitt.edu/obrc/index.php?page=URL1151008585) were then used to predict the potential functions of these target genes in the pathways.
Construction of the lncRNA-microRNA co-expression network
The lncRNA-microRNA regulatory network was defined as described in a previous report (24). The Pearson correlation method was used for miRNA analysis, and those pairs of miRNAs with significant correlations (≥0.90) were chosen to construct the network. Co-expression networks were generated using Cytoscape v.3.10.1 (https://cytoscape.org/); in this representation, each lncRNA corresponded to a node, and the connection of two genes was depicted by an edge, indicating a strong correlation (i.e., either positive or negative).
Statistical analysis
SPSS version 22.0 software (IBM Corp.) was used to analyze the data. Continuous variables were presented as the mean ± SD, or as the median (interquartile range). For each subject, >2-fold changes in the expression of distinct lncRNAs were calculated by dividing the standardized expression levels of the lncRNAs. Differences in the expression levels of the lncRNAs between the HS groups and the healthy controls, with at least 5 biological replicates/group, were analyzed using unpaired Student's t-test, and P<0.05 was considered to indicate a statistically significant value.
Results
Profiling of circulating lncRNAs from patients with HS and healthy controls
The identification of circulating lncRNA signatures in patients with HS was performed in a pilot study (unpublished data). The aim was to identify any significant changes in lncRNA expression in the blood samples of patients with HS and non-HS participants. LncRNA arrays were performed using RNA extracted from the total population of white cells. A 2-fold expression difference was used as the cut-off, and a total of 133 lncRNA transcripts were found to be dysregulated (109 lncRNA transcripts were upregulated, and 24 lncRNA transcripts were downregulated; each P<0.05) in patients with HS compared with non-HS participants (Fig. 1). Using high-signal intensity and ≥6-fold deregulation of gene expression as filters, eight lncRNA candidates were selected. Only three of these eight lncRNA candidates (namely, lncRNA-TINCR, lncRNA-RBM5-AS1 and lncRNA-MRPL23-AS1) were found to be consistently dysregulated in the cohort of blood samples (25 patients with HS and 25 healthy non-HS control participants). The expression levels of circulating lncRNA-TINCR lncRNA-RBM5-AS1 and lncRNA-MRPL23-AS1 in patients with HS were found to be significantly increased compared with non-HS participants (Fig. 2; P=0.004, P=0.002 and P=0.002, respectively).
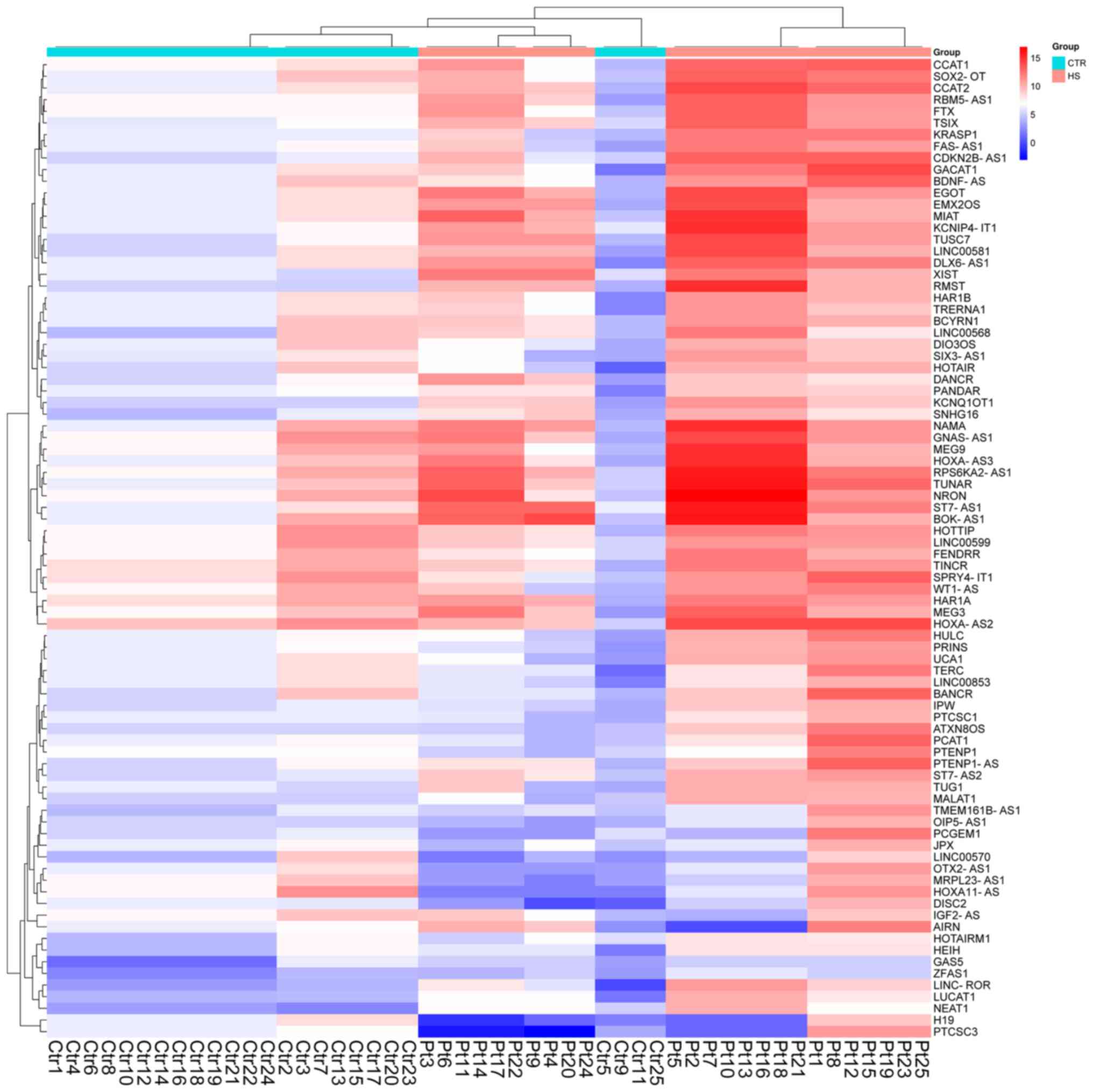

Expression patterns of the circulating lncRNA-TINCR, lncRNA-RBM5-AS1 and lncRNA-MRPL23-AS1 signatures in the blood of patients with HS
As shown in Fig. 2, the expression levels of these three lncRNAs in the peripheral blood was consistently found to be higher in HS blood compared with that of the healthy participants; therefore, the origin of the total RNA was investigated. Further analysis of the data showed that no dysregulated molecules appeared to be sex specific (data not shown). The large standard error values observed for the other lncRNAs is possibly due to the grouping of the samples, given the inherent variability in expression levels among those groups. It is noteworthy that a larger standard error was associated with small expression values, which may be less reproducible. In fact, at low levels of expression, minor technical variations or biological fluctuations can result in relatively large proportional changes, making it challenging to consistently measure these small quantities accurately across samples; however, the absence of dysregulation of these lncRNAs is clear for these groups.
Gene enrichment and ingenuity pathway analysis (IPA) of the lncRNA-co-expressed mRNA signatures
Gene enrichment analysis was performed to determine which genes were involved in the GO categories of biological process, cellular component and molecular function. As shown in Fig. 3, the most enriched GO terms, targeted by lncRNA-co-expressed mRNAs, were found to be responses to regulation of biological and cellular processes (GO: Biological process), cellular and anatomical entities (GO: Molecular function) and intracellular anatomical structures (GO: Cellular component). Examples of relevant functional terms are ‘Cytokine receptor activity’, ‘Non-coding RNA processing’ and ‘Histone deacetylase complex’.

KEGG pathway analysis revealed an involvement of lncRNA-co-expressed mRNAs in pathways such as ‘Toll receptor signaling pathway’, ‘Cadherin signaling pathway’, ‘RHOA signaling’, ‘CCKR signaling pathway’ and ‘ERK5 signaling pathway’ (Fig. 4).

Expression patterns of Yes-associated protein 1 (YAP1), MYC1, SEPTIN7 and interleukin-13 (IL-13) genes from the blood of patients with HS
IPA of lncRNA-co-expressed mRNAs revealed that MYC appeared in several of the pathways associated with the lncRNAs co-expressed mRNAs, followed by YAP1 and SEPTIN7. To gain an understanding of the involvement of these genes in HS etiology, RT-qPCR was performed to measure the expression level of these targets. Both SEPTIN7 and IL-13 were found to be upregulated in the blood of patients with HS, whereas YAP1 and MYC1 exhibited significantly lower expression levels compared with healthy controls (Fig. 5). Taken together, these results suggested that such mRNAs in association with lncRNAs may be involved in the inflammatory state, the maintenance of cellular morphology and tissue development, or the progression of HS.

Construction of the lncRNA/miRNA co-expression network
The lncRNA-miRNA co-expression network was constructed using Cytoscape software (Fig. 6), and the potential interactions between miRNAs and lncRNAs were investigated. LncRNA-TINCR, lncRNA-RBM5-AS1 and lncRNA-MRPL23-AS1 were found to interact with 145 miRNAs, and these were involved in three relevant pathways: i) RhoA signaling and signaling by Rho family GTPases involving Septin3 and Septin7; ii) glucocorticoid receptor signaling/STAT3 and; iii) macrophage alternative activation signaling. Among these pathways, the RhoA signaling and signaling by Rho family GTPases involving Septin3 and Septin7 appears to be the most enriched one. The network indicated that each lncRNA was associated with a large number of target miRNAs, suggesting an inter-regulation of lncRNAs and miRNAs only in the HS state.

Discussion
At present, knowledge on the specific roles of lncRNAs in different types of human disease remains limited. In the present study, 133 dysregulated lncRNAs were identified in the blood samples of patients with HS compared with non-HS participants (fold change ≥2.0; P<0.05), and using RT-qPCR, it was shown that the expression levels of three of the lncRNAs, namely lncRNA-TINCR, lncRNA-RBM5-AS1 and lncRNA-MRPL23-AS1, were significantly upregulated in patients with HS.
In spite of this finding, future studies with larger sample sizes are required to confirm the roles of such candidate lncRNAs in HS. Several functions of lncRNAs can be predicted through the identification of co-expressed mRNAs. In the present study, 45 mRNAs were found to interact with these three lncRNAs in 13 significant pathways (Fig. 4 and Table II). The most enriched network was found to be RhoA Signaling and Signaling by Rho Family GTPases involving the septins, Septin3 and Septin7. There are 13 mammalian septins that are assigned to four groups (Septin2, Septin3, Septin6 and Septin7). Septins have been shown to interact with the membrane and cortical actin cytoskeleton, which partly explains their functions in the cells of the hematopoietic lineages (24). Septins are considered the fourth component of the cytoskeleton, especially the Septin7 isoform, which has a critical role in the formation of higher-order structures. Septin7 may be involved in muscle regeneration and development following muscle injury (25). Preferentially, septins of the Septin3 and Septin7 groups have interactions (26–28).
The results of the present study suggested that there is an inter-regulation of lncRNAs and mRNAs, involved in the cytokinesis, the maintenance of cellular morphology and tissue development, and/or progression of HS. The epithelial adherens junction signaling pathway mainly involves the protein, YAP1 (29). In endothelial cells, the pairing of proteins YAP/transcriptional coactivator with PDZ-binding motif (TAZ) regulates cell decisions made between survival and proliferation or cell death in response to stress (29). YAP/TAZ not only regulates the cell choice in response to mechanical stimuli, but it may also act to reinforce the cytoskeleton and contractile apparatus as a response to mechanical stress. Generally speaking, the YAP/TAZ proteins are overexpressed in different types of cancer, causing hyperproliferation (29). Hyperproliferation of adherens junctions is due to activation or inhibition of YAP/TAZ, depending on the cell density (29).
The glucocorticoid receptor signaling/STAT3 and macrophage alternative activation signaling pathways are important pathways that feature mRNAs linked with lncRNAs. In particular, IL-13 is the gene mainly involved in such pathways. IL-13 is a growth-promoting cytokine that is capable of supporting cell activities such as cell growth, differentiation and apoptosis (30). In atopic dermatitis, an inflammatory and chronic skin disease characterized by an acute level of spongiosis in the basal epidermal layer, IL-13 has been shown to be dysregulated (31). In spongiosis, the skin exists in a morphologically altered state due to the expansion of skin tissue generated by changes in cohesion between epidermal keratinocytes in human skin (32).
The main driver of pathogenesis in HS is an overactivation of immune-cell infiltration, which leads to an enhancement of the inflammatory response due to the release of pro-inflammatory cytokines and chemokines. The inflammation may become chronic and, in severe cases, adversely affects diverse tissues and organs (33). Currently, the etiopathogenesis of HS remains poorly understood due to the complexity and the multifactorial nature of the disease. In the present study, the lncRNA-mRNA interactions were thoroughly identified and explored, and it was shown that altered gene regulation in the lncRNA-mRNA network may be crucial for the development of HS. However, it must be admitted that the present study had numerous limitations associated with the small sample size for microarray analysis and the absence of in vitro and in vivo models to confirm the identified pathways. Specifically, the small sample size not only limited the statistical power of the analysis but also increased the likelihood of encountering large standard errors, particularly for lncRNAs with low expression levels. This can lead to challenges in the reproducibility and accuracy of the present findings, as minor technical or biological variations can significantly affect the measured expression levels of these low-abundance transcripts Therefore, a larger sample cohort, together with experimental validation studies, are required in order to verify the recognized pathways associated with HS.
Acknowledgements
Dr Giorgio Giurato of the University of Salerno (Baronissi, Italy) is acknowledged for his contribution to the analysis and acquisition of the bioinformatics data. Dr Federica Farinella of the University of Campania Luigi Vanvitelli (Caserta, Italy) is acknowledged for their contribution to the availability of the data and for the final revision process.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be found in the ArrayExpress database under accession number E-MTAB-13699 or at the following URL: (https://www.ebi.ac.uk/biostudies/arrayexpress/studies?query=E-MTAB-13699).
Authors' contributions
BDF, AD and EN performed the data analysis. RS performed RNA extraction. PDL, MM, CM and EN performed the microarray assay and RT-qPCR expression analysis of patient samples. GB, GA, GR and EM recruited the patients and collected clinical data. BDF was the principal investigator. All authors read and approved the final version of the manuscript. BDF and AD confirm the authenticity of all the raw data.
Ethics approval and consent to participate
The research protocol was conducted under the guidelines of The Ethics Committee of the University of Campania Luigi Vanvitelli, who also approved the research (approval no. 12478/20; Caserta, Italy). The samples were handled according to the guidelines of the Helsinki Declaration. Written informed consent was obtained from all patients and healthy controls prior to enrolment.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
|
Mattick JS, Amaral PP, Carninci P, Carpenter S, Chang HY, Chen LL, Chen R, Dean C, Dinger ME, Fitzgerald KA, et al: Long non-coding RNAs: Definitions, functions, challenges and recommendations. Nat Rev Mol Cell Biol. 24:430–447. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Schmitz SU, Grote P and Herrmann BG: Mechanisms of long noncoding RNA function in development and disease. Cell Mol Life Sci. 73:2491–2509. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Kazimierczyk M, Kasprowicz MK, Kasprzyk ME and Wrzesinski J: Human Long Noncoding RNA Interactome: Detection, characterization and function. Int J Mol Sci. 21:10272020. View Article : Google Scholar : PubMed/NCBI | |
|
Herman AB, Tsitsipatis D and Gorospe M: Integrated lncRNA function upon genomic and epigenomic regulation. Mol Cell. 82:2252–2266. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Devaux Y, Zangrando J, Schroen B, Creemers EE, Pedrazzini T, Chang CP, Dorn GW II, Thum T and Heymans S; Cardiolinc network, : Long noncoding RNAs in cardiac development and ageing. Nat Rev Cardiol. 12:415–425. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Liu Y, Zhang Y, Chen C and Li Y: lncRNA HIF1A-AS2: A potential oncogene in human cancers (Review). Biomed Rep. 15:852021. View Article : Google Scholar : PubMed/NCBI | |
|
Chen M, Lei N, Tian W, Li Y and Chang L: Recent advances of non-coding RNAs in ovarian cancer prognosis and therapeutics. Ther Adv Med Oncol. 14:175883592211180102022. View Article : Google Scholar : PubMed/NCBI | |
|
Jiang XY, Zhu QC, Zhang XJ, Duan T, Feng J, Sui XB, Sun XN and Mou YP: Roles of lncRNAs in pancreatic ductal adenocarcinoma: Diagnosis, treatment, and the development of drug resistance. Hepatobiliary Pancreat Dis Int. 22:128–139. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Ashekyan O, Abdallah S, Shoukari AA, Chamandi G, Choubassy H, Itani ARS, Alwan N and Nasr R: Spotlight on Exosomal Non-Coding RNAs in breast cancer: An in silico analysis to identify potential lncRNA/circRNA-miRNA-Target Axis. Int J Mol Sci. 23:83512022. View Article : Google Scholar : PubMed/NCBI | |
|
Hao Y, Xie B, Fu X, Xu R and Yang Y: New Insights into lncRNAs in Aβ Cascade Hypothesis of Alzheimer's Disease. Biomolecules. 12:18022022. View Article : Google Scholar : PubMed/NCBI | |
|
Ghafouri-Fard S, Poulet C, Malaise M, Abak A, Mahmud Hussen B, Taheriazam A, Taheri M and Hallajnejad M: The emerging role of non-coding RNAs in osteoarthritis. Front Immunol. 12:7731712021. View Article : Google Scholar : PubMed/NCBI | |
|
Sabaie H, Salkhordeh Z, Asadi MR, Ghafouri-Fard S, Amirinejad N, Askarinejad Behzadi M, Hussen BM, Taheri M and Rezazadeh M: Long Non-Coding RNA- Associated Competing Endogenous RNA Axes in T-Cells in multiple sclerosis. Front Immunol. 12:7706792021. View Article : Google Scholar : PubMed/NCBI | |
|
De Benedittis G, Ciccacci C, Latini A, Novelli L, Novelli G and Borgiani P: Emerging Role of microRNAs and long non-coding RNAs in Sjögren's Syndrome. Genes (Basel). 12:9032021. View Article : Google Scholar : PubMed/NCBI | |
|
Sirbe C, Simu G, Szabo I, Grama A and Pop TL: Pathogenesis of autoimmune hepatitis-cellular and molecular mechanisms. Int J Mol Sci. 22:135782021. View Article : Google Scholar : PubMed/NCBI | |
|
Lv J, Liu Y, Cui J, Fang H, Wu Y, Zhu X, Guo M, Li C, Dou J, Chen Z and Du X: Profile screening of differentially expressed lncRNAs of circulating leukocytes in type 2 diabetes patients and differences from type 1 diabetes. Front Endocrinol (Lausanne). 12:6905552022. View Article : Google Scholar : PubMed/NCBI | |
|
Wan DC and Wang KC: Long Noncoding RNA: Significance and potential in skin biology. Cold Spring Harb Perspect Med. 4:a0154042014. View Article : Google Scholar : PubMed/NCBI | |
|
Chu CB, Yang CC and Tsai SJ: Hidradenitis suppurativa: Disease pathophysiology and sex hormones. Chin J Physiol. 64:257–265. 2021. View Article : Google Scholar : PubMed/NCBI | |
|
Fabbrocini G, Ruina G, Giovanardi G, Dini V, Raone B, Venturini M, Caposiena Caro RD, Veraldi S, Cannavò SP, Merlo G, et al: Hidradenitis suppurativa in a large cohort of Italian patients: Evaluation of the Burden of disease. Dermatology. 238:487–497. 2022. View Article : Google Scholar : PubMed/NCBI | |
|
Scala E, Cacciapuoti S, Garzorz-Stark N, Megna M, Marasca C, Seiringer P, Volz T, Eyerich K and Fabbrocini G: Hidradenitis Suppurativa: Where we are and where we are going. Cells. 10:20942021. View Article : Google Scholar : PubMed/NCBI | |
|
Nigro E, Polito R, Babino G, Mattera E, Fulgione E, Ragozzino G, D'Esposito V, Cabaro S, Signoriello G, Formisano P, et al: Adiponectin contributes to the inflammatory milieu in hidradenitis suppurativa. Dermatol Pract Concept. 12:e20221572022. View Article : Google Scholar : PubMed/NCBI | |
|
Garg A, Naik HB, Alavi A, Hazen P, Hsiao JL, Shi VY, Weisman J, Tran T, Rudnik J, Jedrzejczyk A, et al: Real-World findings on the characteristics and treatment exposures of patients with hidradenitis suppurativa from US claims data. Dermatol Ther (Heidelb). 13:581–594. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Rao X, Huang X, Zhou Z and Lin X: An improvement of the 2ˆ(−delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat Bioinforma Biomath. 3:71–85. 2013.PubMed/NCBI | |
|
Livak KJ and Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI | |
|
Yu G, Yao W, Wang J, Ma X, Xiao W, Li H, Xia D, Yang Y, Deng K, Xiao H, et al: LncRNAs expression signatures of renal clear cell carcinoma revealed by microarray. PLoS One. 7:e423772012. View Article : Google Scholar : PubMed/NCBI | |
|
Kim J, Mooren OL, Onken MD and Cooper JA: Septin and actin contributions to endothelial cell-cell junctions and monolayer integrity. Cytoskeleton (Hoboken). 80:228–241. 2023. View Article : Google Scholar : PubMed/NCBI | |
|
Nakahira M, Macedo JN, Seraphim TV, Cavalcante N, Souza TA, Damalio JC, Reyes LF, Assmann EM, Alborghetti MR, Garratt RC, et al: A draft of the human septin interactome. PLoS One. 5:e137992010. View Article : Google Scholar : PubMed/NCBI | |
|
Neubauer K and Zieger B: The mammalian septin interactome. Front Cell Dev Biol. 5:32017. View Article : Google Scholar : PubMed/NCBI | |
|
Gönczi M, Ráduly Z, Szabó L, Fodor J, Telek A, Dobrosi N, Balogh N, Szentesi P, Kis G, Antal M, et al: Septin7 is indispensable for proper skeletal muscle architecture and function. Elife. 11:e758632022. View Article : Google Scholar : PubMed/NCBI | |
|
Dasgupta I and McCollum D: Control of cellular responses to mechanical cues through YAP/TAZ regulation. J Biol Chem. 294:17693–17706. 2019. View Article : Google Scholar : PubMed/NCBI | |
|
Kim K, Kim H and Sung GY: An Interleukin-4 and Interleukin-13 Induced Atopic Dermatitis Human Skin Equivalent Model by a Skin-On-A-Chip. Int J Mol Sci. 23:21162022. View Article : Google Scholar : PubMed/NCBI | |
|
Napolitano M, di Vico F, Ruggiero A, Fabbrocini G and Patruno C: The hidden sentinel of the skin: An overview on the role of interleukin-13 in atopic dermatitis. Front Med (Lausanne). 10:11650982023. View Article : Google Scholar : PubMed/NCBI | |
|
De Vuyst E, Salmon M, Evrard C, Lambert de Rouvroit C and Poumay Y: Atopic Dermatitis Studies through in vitro models. Front Med (Lausanne). 4:1192017. View Article : Google Scholar : PubMed/NCBI | |
|
De Felice B, Montanino C, Mallardo M, Babino G, Mattera E, Ragozzino G, Argenziano G, Daniele A and Nigro E: Circulating microRNAs in Hidradenitis Suppurativa. Genes (Basel). 13:15442022. View Article : Google Scholar : PubMed/NCBI |