Neuroinflammation is a response orchestrated by the
central nervous system (CNS) in response to infection and injury.
The acute neuroinflammatory response reduces damage by promoting
the repair of injured tissue. However, persistent stimulation leads
to the transformation of the inflammatory response from acute to
chronic, resulting in neuronal functional impairment and thus
facilitates the progression of CNS diseases (1). Chronic neuroinflammation has been
reported as a pathological feature present in several
neurodegenerative diseases such as Alzheimer's disease (AD),
Parkinson's disease (PD), multiple sclerosis (MS) and epilepsy,
amongst other neurological disorders (2,3).
The close association between neuroinflammation and
neurodegeneration suggests that neuroinflammatory mechanisms may
trigger neuronal degeneration, leading to neurotoxicity and a loss
of neuronal cells.
Microglia, which are activated by pathological
stimuli such as infections, foreign pathogens and
neurodegeneration, produce chemotactic factors and proinflammatory
cytokines, including nitric oxide, reactive oxygen species (ROS),
interleukin (IL)-1β, IL-6 and tumor necrosis factor-α (TNF-α), to
eliminate detrimental elements. Nonetheless, persistent stimulation
can lead to an overabundance of inflammatory factors, which in turn
inflict damage upon neurons (4).
Likewise, astrocytes exhibit dual roles in neuroinflammation. In
response to cerebral trauma, astrocytes undergo proliferation and
transition into a neuroprotective state, fostering reparative and
regenerative mechanisms such as remyelination (5). Conversely, in neuroinflammatory
diseases, astrocytes are excessively activated into a neurotoxic
state by cytokines secreted by microglia, releasing uncontrolled
pro-inflammatory cytokines and complement proteins, which
exacerbate damage to neighboring cells (6,7).
Moreover, astrocytes facilitate lymphocyte movement across the
blood-brain barrier, engage in antigen presentation between
lymphocytes and microglia, and activate peripheral B cells and T
cells via the lymphatic system, thereby amplifying the cerebral
inflammatory cascade (1,8,9).
In the CNS, mitochondria serve as the primary energy
source for cellular metabolic processes and are pivotal in
regulating cellular metabolism, calcium signaling and programmed
cell death (10). Neurons rely
on mitochondrial oxidative phosphorylation (OXPHOS) to meet their
energy demands, maintain ion gradients and facilitate
neurotransmitter uptake and recycling (11). Astrocytes contribute to the
mitigation of neuronal and oligodendrocyte free fatty acid
peroxidation and ROS generation through mitochondrial fatty acid
β-oxidation (FAO), which accounts for ~20% of the brain's energy
supply (12). Additionally,
mitochondrial calcium ions modulate key enzymes of the
tricarboxylic acid cycle, such as pyruvate dehydrogenase, thereby
regulating OXPHOS (13).
Mitochondrial calcium ions also regulate synaptic communication and
excitability by modulating astrocytic proliferation and the release
of excitotoxic glutamate (14).
In addition, microglial mitochondria enhance migration and
phagocytosis through calcium ion influx (15). Once mitochondrial dysfunction
occurs, the mitochondria become insufficient to meet the heightened
energy demands of overstimulated neurons and hyperactive glial
cells, leading to abnormal cell metabolism and widespread cell
death (10).
Mitochondrial dysfunction serves as both a cause and
a consequence of chronic neuroinflammatory diseases. Neuronal
mitochondrial dysfunction has been observed in AD, PD and
amyotrophic lateral sclerosis (16-18). Chronic inflammation leads to the
secretion of cytokines that sustain inflammation and redox stress,
inducing mitochondrial DNA (mtDNA) damage (19). Correspondingly, damaged
mitochondria can further induce persistent inflammatory responses
and downstream pathological inflammation (20,21). A study has shown that inhibiting
mitochondrial complex I activates microglia, whereas inhibiting
mitochondrial fission reduces pro-inflammatory cytokine generation
(12). Due to mitochondrial
damage, overactivated microglia undergo a metabolic shift from
OXPHOS to glycolysis, resulting in increased generation of ROS and
reactive nitrogen species (RNS), thereby exacerbating the
inflammatory response (22).
Concurrently, microglia can induce the generation of
pro-inflammatory astrocytes by releasing fragmented mitochondria
(23). Furthermore, impairment
of mitochondrial FAO in astrocytes contributes to the development
of neuroinflammation and subsequent neurodegenerative processes
(24). Additionally, the
accumulation of damaged mitochondria in neurons can accelerate the
progression of diseases by initiating programmed cell death
(23). Mitochondria may
therefore be a key link between chronic neuroinflammation and the
pathogenesis of neurodegenerative diseases. Thus, repairing
mitochondrial dysfunction may improve the outcomes of
neurodegenerative diseases, such as AD and PD (25,26).
The aim of the present review was to summarize the
molecular characteristics of mitochondrial dysfunction and provide
potential directions for targeting mitochondria in the treatment of
chronic neuroinflammatory diseases.
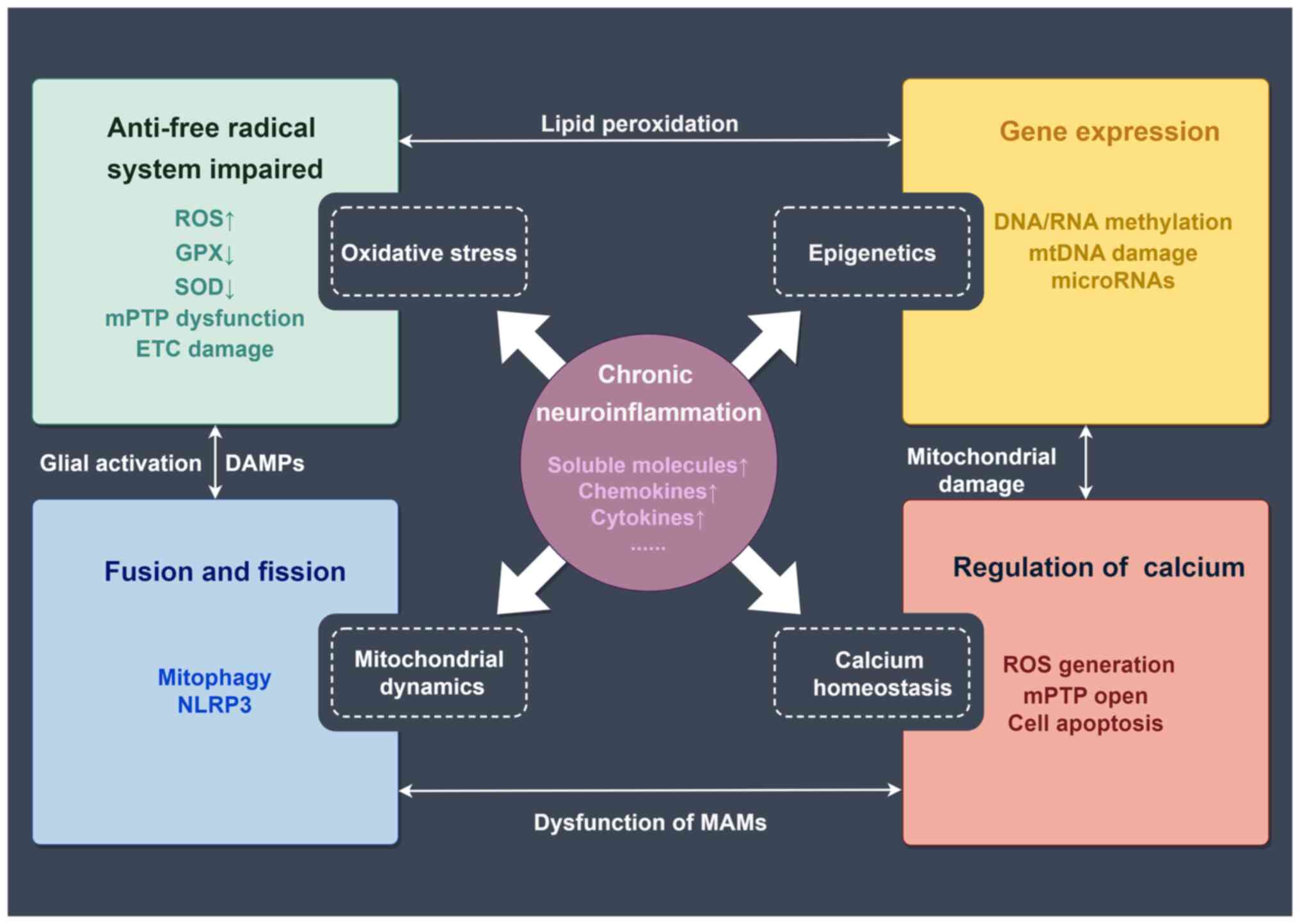
Mechanisms of mitochondrial dysfunction involved in
the progression and prognosis of chronic neuroinflammatory diseases
include oxidative stress, epigenetics, mitochondrial dynamics and
calcium homeostasis (27-32)
(Fig. 1).
In normal, healthy cells, 90% of ROS are generated
as a result of cellular respiration. During this process, electrons
detach from the electron transport chain and attach to oxygen,
producing superoxide anions (O2−) (33). Additionally, metal enzymes
present within organisms utilize the interaction between oxygen and
metal ions to generate ROS, which is a result of cellular
metabolism (34). Conversely,
normal cells also possess a protective system against free
radicals, primarily composed of antioxidant enzymes such as
glutathione peroxidase (GPX), non-enzymatic antioxidant factors,
superoxide dismutase (SOD) and catalase (33). The excessive reduction of free
radicals is catalyzed by antioxidant enzymes. SOD acts on
O2− to produce hydrogen peroxide
(H2O2), which has a lower oxidative capacity
than O2−, while catalase and GPX enzymes,
with the assistance of certain cofactors, convert
H2O2 into H2O. When this
regulatory process is disrupted, ROS can inflict destructive damage
on cells (33). Excessive ROS
further induces peroxidation modifications of cellular
macromolecules such as lipids, proteins, RNA and DNA (35). For instance, protein peroxidation
may acquire toxic functions by forming cytotoxic aggregates.
Therefore, the accumulation of ROS caused by various factors (such
as increased oxygen consumption in the brain due to high energy
demand, elevated levels of unsaturated fatty acids in neuronal
membranes, high levels of redox transition metal ions, low
antioxidant levels and neurotransmitter oxidation) makes the brain
highly susceptible to the damaging effects of oxidative stress
(36). The excessive generation
of ROS, leading to oxidative stress, has emerged as a shared
underlying mechanism implicated in multiple chronic
neuroinflammatory disorders, such as AD and PD (37,38).
Due to being the primary source of ROS,
mitochondrial dysfunction appears to be a potential focal point for
the underlying pathology of neuroinflammation (39). Excessive free radicals damage the
inner mitochondrial membrane, leading to compromised mitochondrial
energy production and metabolism in the brain. This results in
neuronal dysfunction and further exacerbates oxidative stress,
promoting neuronal dysfunction and apoptosis. Furthermore, free
radicals can directly or indirectly induce abnormal mitochondrial
permeability transition pore (mPTP) function, indirectly altering
the fluidity, permeability and osmotic properties of the
mitochondrial membrane, thereby facilitating mPTP-related ROS
release (40). Moreover, free
radicals also interfere with electron transport chain (ETC)
complexes, further promoting ROS generation (41). A study has also reported that
mitochondrial ROS (mtROS) can lead to impairment of complex I
within the mitochondrial ETC. This in turn results in a reduction
of mitochondrial OXPHOS efficiency (42). This cycle formed by mitochondrial
dysfunction and inflammation-related oxidative stress exacerbates
the pathological damage in neuroinflammatory disorders.
Epigenetic modifications within the mitochondria can
influence mitochondrial gene expression and function. In
neuroinflammatory conditions, heightened ROS production induces
deleterious effects on mitochondrial respiration and OXPHOS,
leading to DNA oxidation, rearrangements and mutations (43). mtDNA, with its elevated mutation
rate and proximity to OXPHOS sites, is more susceptible to
oxidative stress compared with nuclear DNA (43). Methylation is a primary
epigenetic mechanism within mitochondria due to the absence of
histones in mtDNA (44).
Decreased mtDNA methylation levels have been observed in blood
samples and postmortem brain tissues from individuals with
neuroinflammatory diseases (44). Additionally, chronic stress
activates the hypothalamic-pituitary-adrenal axis, resulting in
excessive glucocorticoid release and the regulation of mtDNA
transcription and mtRNA expression (45). Conversely, mtDNA mutations
exacerbate ROS production and trigger apoptosis through disruptions
in the electron transport chain, impaired protein synthesis, and
increased replication errors, influencing disease onset and
progression (46). MtDNA damage
results in impaired ETC function, reduced ATP generation, increased
levels of ROS and disrupted calcium homeostasis, leading to
exacerbated amyloid-β (Aβ) processing and aggregation in AD mice
(47). Another study reported
that Aβ induced mtDNA methylation, which persisted after the
removal of Aβ and induced cognitive impairment in AD (48). Furthermore, non-coding RNAs are
implicated in chronic low-grade systemic inflammation, known as
inflammageing, impacting the energetic, oxidative and inflammatory
status of senescent cells by modulating NF-κB/NLR family pyrin
domain containing 3 (NLRP3) pathways and triggering
senescence-associated secretory phenotype (43). Downregulation of microRNAs could
also promote neuroinflammation by affecting the expression of genes
critical for neuronal function and immune response in PD (49). It is evident that mitochondrial
epigenetics is closely associated with the development of
neuroinflammation. Understanding the regulatory role of
mitochondrial epigenetics is therefore crucial for unraveling the
underlying mechanisms of various neurological disorders.
The complex processes of mitochondrial fusion and
fission collectively maintain mitochondrial functionality in the
face of cellular metabolic or environmental stress (50). Mitochondrial fusion involves the
merging of individual mitochondria, resulting in larger and
interconnected networks. This process allows for the exchange of
contents, including proteins, lipids and mtDNA, thereby promoting
functional complementation and maintaining mitochondrial integrity.
Mitochondrial fission is the opposite of fusion and involves the
division of mitochondria into smaller fragments. Fission has a
crucial role in quality control mechanisms, as it allows for the
removal of damaged or dysfunctional portions of mitochondria
through a process termed 'mitophagy' (51). Moreover, mitochondrial fission
also facilitates the distribution of mitochondria throughout the
cell (52).
Chronic neuroinflammation disrupts mitochondrial
dynamics. Under neuroinflammatory conditions, dysfunctional
mitochondria release ROS and damage-associated molecular patterns
(DAMPs) (53), activating
microglia and astrocytes, and triggering the release of
pro-inflammatory cytokines and chemokines (54), thereby exacerbating the damage.
In addition, disruption of mitochondrial dynamics can result in the
accumulation of dysfunctional mitochondria, leading to increased
susceptibility to inflammation-induced neuronal death. For
instance, microglia can activate astrocytes into a neurotoxic state
by releasing mitochondrial fragments and damaged mitochondria,
further mediating extracellular neuronal death in neuroinflammation
(23). Furthermore, mitophagy
also proves advantageous in eliminating impaired mitochondria and
decreasing the infiltration of inflammatory molecules at the
location where damaged mitochondria accumulate (55).
Mitochondrial dynamics may also have a significant
involvement in inflammasome activation in chronic inflammation.
Inhibition of dynamin-related protein 1 (Drp1) and overexpression
of fusion proteins can attenuate inflammation-associated
inflammasome responses (56).
During RNA virus infection, mitofusin-2 interacts with NLRP3 to
activate inflammasomes (57).
Thus, molecules involved in mitochondrial dynamics may be crucial
regulators of inflammasome activation. In summary, mitochondrial
dynamics are essential for maintaining neuronal health and survival
and a balanced fusion and fission process is beneficial for
maintaining healthy mitochondrial function.
Mitochondrial calcium homeostasis holds significant
importance in maintaining the functionality of neurons and glial
cells (58). Mitochondrial
function not only sustains the energy prerequisites of both
spontaneous and induced neuronal activities in the brain through
energy metabolism, but also governs neuronal signaling via uptake
and cycling of mitochondrial calcium ions (59). Furthermore, the dynamic
regulation of mitochondrial calcium homeostasis is also important
for cell survival (13).
In the progression of neuroinflammatory diseases,
calcium homeostasis remains a crucial molecular mechanism (60). The dysregulation of neuronal
calcium homeostasis leads to oxidative stress, mitochondrial
dysfunction, protein conversion disorders and neuroinflammation
(61). Activated glial cells
serve a crucial role in neuroinflammation and release soluble
signaling molecules, including chemokines, pro-inflammatory
cytokines, glutamate, prostaglandins, ROS, RNS and damaged
mitochondria (62-64). Astroglial calcium signaling
appears to be dysregulated in AD, which is potentially linked to
the accumulation of Aβ in the brain (65). Depletion of mitochondrial calcium
transporters has been shown to mitigate the inflammatory damage
caused by glial cells activated by lipopolysaccharides (66).
AD is a gradually advancing neurodegenerative
condition characterized by the presence of Aβ and τ protein
tangles, which are considered distinctive pathological markers. Aβ
accumulation has also been observed within the mitochondria in the
brains of patients with AD and transgenic AD mouse models (77,78). Aβ can directly disrupt the ETC
and interfere with various mitochondrial matrix proteins and
putative components of the mPTP, ultimately resulting in
mitochondrial dysfunction (77-80).
In the early stages of AD, another often observed
mitochondrial abnormality is the excessive generation of ROS,
culminating in an upsurge of oxidative stress (81). Oxidative stress causes neuronal
cell death, which contributes to the progressive cognitive decline
seen in AD (82). Normally,
mitochondria serve as pivotal guardians of the cellular redox
equilibrium, orchestrating this balance via their antioxidant
defense systems. However, malfunctioning mitochondria compromise
these protective mechanisms, resulting in diminished scavenging of
ROS and an escalation in oxidative harm (83). Furthermore, surplus ROS within
mitochondria harms lipids and proteins. For instance, lipid
peroxidation engenders the production of harmful byproducts such as
malondialdehyde and 4-hydroxynonenal, intensifying the oxidative
stress milieu (84).
Concomitantly, protein oxidation can induce structural and
functional impairments in mitochondrial proteins, thereby impacting
energy synthesis and overall integrity. It is also worth noting
that the heightened oxidative stress observed in AD can precipitate
mutations, deletions and impairments in mtDNA repair mechanisms
(85). These events further
compound mitochondrial dysfunction, instigating a cycle of
oxidative stress and neuronal damage.
Furthermore, DAMPs released from compromised
mitochondria, coupled with elevated ROS levels, serve to intensify
immune responses, with microglia playing a pivotal regulatory role
in this process (86). On the
one hand, activated microglia contribute to reducing
neuroinflammation by phagocytosing and eliminating Aβ, while on the
other hand, these microglia release pro-inflammatory cytokines and
other inflammatory molecules, thus promoting inflammation (86,87). Notably, emerging research
suggests that there is a bidirectional communication between
mitochondria and microglia (88,89). Damaged mitochondria release mtDNA
fragments into the cytoplasm, which can activate immune responses
through Toll-like receptor 9, NLRP3 and stimulator of interferon
genes (STING) signaling pathways. Microglia recognize these mtDNA
fragments as danger signals and respond by releasing inflammatory
mediators that further amplify the inflammatory microenvironment,
inducing mitochondrial damage and subsequent cell death (89). This communication may also
perpetuate neuroinflammation and contribute to the progression of
AD. In addition, activation of the NLRP3 inflammasome is also an
important factor in the pathogenesis of AD (90,91), leading to the release of potent
pro-inflammatory cytokines such as its effector molecule, IL-1β
(92). Elevated IL-1β levels
have been detected in the serum, cerebrospinal fluid and brain
tissues of patients with AD (93). IL-1β can enhance the neuronal
production of Aβ and induce τ protein phosphorylation, the blocking
of which can alleviate neuroinflammation by reducing Aβ levels and
τ activation (94,95). IL-18, another proinflammatory
cytokine released when the NLRP3 inflammasome is activated, has
been demonstrated to be correlated with susceptibility to sporadic
late-onset AD (92,96). In conclusion, the
neuroinflammatory process plays a crucial role in AD development,
and understanding the intricate relationship between mitochondria
and neuroinflammation in AD offers potential therapeutic avenues
(Fig. 2).
PD is the second most prevalent neurodegenerative
disorder, succeeding AD, with a primary impact on the elderly
population (97). PD is
characterized by the subclinical presence of cytoplasmic
proteinaceous aggregates, specifically α-synuclein, which
congregate to form Lewy bodies (LBs) within the substantia nigra.
This process is coupled with a decline in dopaminergic (DA)
neurons. The extensive degeneration of these DA neurons results in
diminished dopamine levels within the brain, which gives rise to a
spectrum of clinical manifestations, encompassing challenges in
maintaining posture, the emergence of stationary tremors, a
decrease in movement speed (bradykinesia) and the onset of joint
stiffness reminiscent of ankylosing arthritis (49). Following the loss of
dopamine-producing neurons, there is a subsequent degeneration of
other neuronal subtypes, giving rise to symptoms unresponsive to
dopamine modulation. These symptoms encompass a spectrum of
manifestations, including insomnia, compromised olfactory
perception, dysregulation of the autonomic system, pain perception
alterations and sensory dysfunction (98). However, the precise
pathophysiological mechanisms driving PD have remained elusive. In
addition to α-synuclein, there is mounting evidence implicating
other genetic mutations such as in Parkin, leucine-rich
repeat kinase 2 (LRRK2) and DJ-1, alongside
environmental factors (including exogenous neurotoxins, age and
diet) as potential contributors to the etiology of PD (99,100). These factors intricately
contribute to the processes of neurodegeneration and
neuroinflammation stemming from oxidative stress, α-synuclein
oligomerization and mitochondrial dysfunction. Notably, it has been
observed that α-synuclein is also present on the mitochondrial
surface, exerting an impact on mitochondrial structural integrity
and functional dynamics (101).
Impaired complex I has been found in samples from patients with PD
and the introduction of toxins inhibiting complex I has been shown
to lead to the loss of dopaminergic cells and the manifestation of
Parkinson's disease symptoms (102). Furthermore, the presence of
mutations in mtDNA has been identified within neurons of
individuals with PD (103). As
a result, mitochondrial dysfunction emerges as a recurring
determinant in the context of PD.
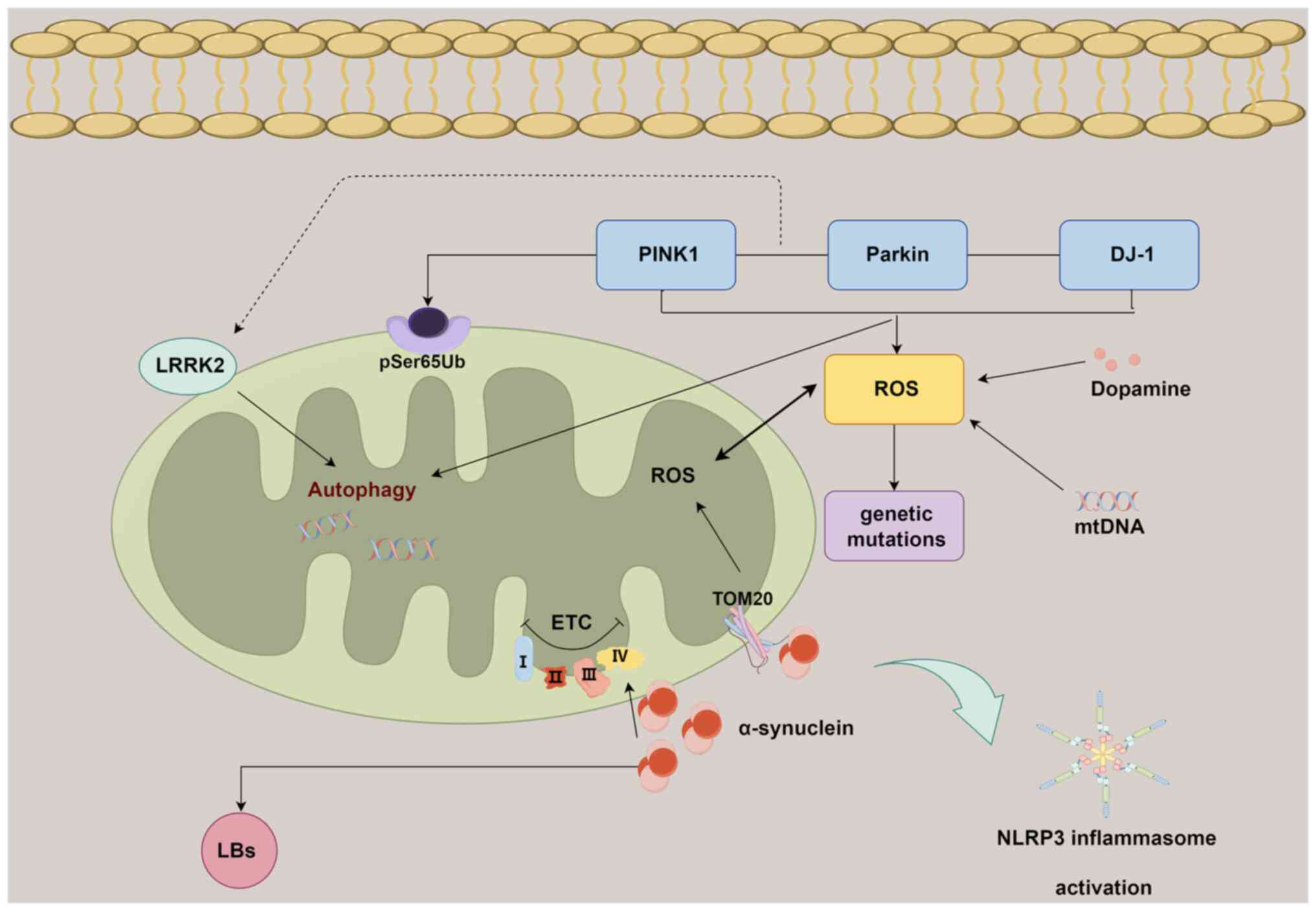
The PTEN-induced kinase 1 (PINK1)/Parkin pathway
serves a pivotal role in the context of mitochondrial dysfunction
and its association with PD. In the current understanding of PD,
mutations within the PINK1 (PARK6) and Parkin (PARK2)
genes are thought to be associated with the manifestation of
autosomal recessive early-onset PD (104). Research has demonstrated that
the PINK1/Parkin pathway participates in the progression of PD by
influencing mitochondrial autophagy (102,103,105). While mice lacking PINK1 or
Parkin do not exhibit significant PD-related phenotypes, a study
has demonstrated that these mice accumulate mtDNA mutations, which
consequently promotes inflammation in aged Parkin−/−
(also termed 'Mutator') mice. This pathological progression appears
to be modulated by STING signaling (106). Furthermore, elevated levels of
phosphorylated serine 65 of ubiquitin and PRKN have been observed,
which are associated with the phosphorylation of ubiquitin by PINK1
at the outer mitochondrial membrane (OMM), have been identified in
postmortem PD brains (107).
These investigations substantiate a notable association between
neuroinflammation and the activation of the PINK1/Parkin pathway in
PD, indicating that mitochondrial autophagy has a pivotal role in
averting neuroinflammation within this pathological framework.
The involvement of α-synuclein in perturbing
mitochondrial function has been previously substantiated (108). It has been documented that
α-synuclein possesses a mitochondrial targeting sequence at its
N-terminal region, allowing its localization to the OMM. This
localization facilitates interactions with components of the outer
membrane receptors, thereby leading to compromised cellular
respiration (109). This
phenomenon has been observed in models of PD and in post-mortem
brain tissue from individuals with PD (108,110). Certain α-synuclein species have
the capability to intricately bind with the translocase of outer
mitochondrial membrane 20 receptor, contributing to mitochondrial
dysfunction and an elevated generation of ROS (108). Notably, a study employed a
seeding-based model of α-synuclein fibrillization to validate that
the progression of LB formation, beyond mere fibril assembly, is a
key catalyst in neurodegeneration, additionally exacerbating
mitochondrial impairment and synaptic dysfunction (111). This suggests an intrinsic link
between mitochondrial dysfunction, α-synuclein aggregation and the
formation of LBs.
The NLRP3 inflammasome assumes a pivotal role in
instigating the neuroinflammatory cascade observed in PD.
Heightened levels of inflammasome constituents and
inflammation-associated factors have been discerned within blood
samples sourced from individuals with PD (105,115). Mitochondrial impairment within
microglia, coupled with the activation of the NLRP3 inflammasome,
has been reported in both in vitro and in vivo models
of PD (116). In addition,
activation of NLRP3 has been observed in PINK1−/− or
Parkin−/− microglia, while inhibitors of the
inflammasome can effectively suppress this activation process
(117). Furthermore, the
attenuation of NLRP3 inflammasome activation not only mitigates
neuroinflammation and ameliorates motor impairments but also
safeguards against the depletion of DA neurons in both a
mPTP-induced PD model and a human α-synuclein overexpression PD
model (118).
Collectively, the convergence of α-synuclein
oligomerization, genetic mutations, impaired mitochondrial
autophagy and NLRP3 activation constitutes a synergistic interplay
contributing to mitochondria-associated neuroinflammation during
the progression of PD (Fig.
3).
MS is a chronic inflammatory disease of the CNS
characterized by demyelination and axonal degeneration (119). The inflammation observed in MS
arises from elements of both the innate and adaptive immune
systems, encompassing the proliferation and dysregulation of
pro-inflammatory T lymphocytes, activation of B cells and secretion
of inflammatory cytokines (120). At the onset of MS, pathogenic
inflammatory T lymphocytes infiltrate the CNS, triggering an immune
response that activates microglia and astrocytes, leading to acute
inflammation. Subsequently, B cells are further activated,
initiating a cascade that sustains chronic inflammation (121). While anti-inflammatory and
immunomodulatory therapies have become mainstream in the treatment
of acute demyelinating episodes, options remain limited for
addressing the progressive stages of MS (122). Further exploration of the
pathogenesis of MS is therefore still required.
A recent study has substantiated that mitochondrial
dysfunction contributes to CNS damage in MS (123). Mitochondria function as the
principal energy supply units within neurons. Neurons facilitate
signal transmission through membrane depolarization, which is
facilitated by the electrochemical gradient of
Na+/K+-ATPase. In the context of MS, the
interplay of chronic inflammation and myelin disruption leads to a
redistribution of ion channels. The heightened presence of
Na+/K+-ATPase intensifies ATP consumption
(123). At this critical
juncture, mitochondria can compensate by augmenting both their
quantity and volume, thereby inducing alterations in neuron
positioning and morphology (123). Persistent inflammation triggers
the activation of macrophages and microglial cells, thereby
instigating the release of ROS and inducing oxidative stress
(124). This exacerbates the
release of glutamate, ultimately culminating in neuronal damage
(120). Oxidative stress
imposes secondary damage on both mitochondria and macromolecules
(such as mtDNA, ETC proteins and lipids), thereby significantly
impairing energy generation (120). While nuclear factor erythroid
2-related factor 2 and antioxidant enzymes such as heme
oxygenase-1, are activated during periods of hypoxic stress to
compensate for mitochondrial dysfunction, once a critical threshold
of reduced ATP production is reached, ion homeostasis becomes
compromised (125). This
disruption results in chronic inflammation and triggers
Ca2+-dependent proteases, ultimately leading to
apoptosis within demyelinated axons (126). The presence of oxidized DNA and
lipids has been observed in apoptotic oligodendrocytes and
dystrophic axons within active MS lesions (127). Furthermore, the inflammatory
factor, TNF-α, exacerbates the impairment of OXPHOS through
Ca2+ modulation (128). The resultant reduction in ATP
production hampers the ability of the
Na+/K+-ATPase to maintain gradients after
action potentials, leading to an accumulation of sodium within the
neuronal cytoplasm. This phenomenon, in turn, compels
Na+/Ca2+channels to facilitate intracellular
calcium transfer, initiating a cascade of Ca2+-dependent
apoptosis that ultimately culminates in neuronal death. This
intricate process significantly contributes to Wallerian
degeneration and irreversible neurofunctional impairment (120). In MS animal models,
double-strand breaks in mtDNA lead to chronic demyelination and
axonal degeneration, which are exacerbated over time (129). It is noteworthy that
mitochondrial dysfunction and oligodendrocyte myelin formation are
inherently interconnected. The level of the mitochondrial
metabolite, N-acetylaspartate (NAA), is reduced in the
normal-appearing white matter of patients with MS (130,131). In vitro experiments have
confirmed that extracellular NAA improves Oli-neuM cell
differentiation and axonal connectivity (132). Furthermore, the NLRP3
inflammasome and cyclic GMP-AMP synthase-STING pathway, which are
associated with increased mitochondrial damage and respiratory
stress, are activated in MS (133,134). These findings highlight the
pivotal role of mitochondrial function in the progression of MS
(Fig. 4).
Epilepsy is a persistent neurological condition
distinguished by the recurrence of seizures intertwined with an
underlying neurodegenerative process (135). The intricate diversity of
epilepsy presents a significant challenge for its treatment
(136). Moreover, the success
rate of antiepileptic drug therapy remains limited, ranging from 30
to 50% (137). Lately, there
has been growing interest in the role of oxidative stress and redox
dysregulation in epilepsy. Elevated levels of diverse biomarkers
associated with oxidative stress and neuroinflammation have been
reported in the brains and peripheral tissues of both human
patients and animal epilepsy models (138,139). Therefore, anti-inflammatory and
antioxidant therapies hold promising therapeutic potential.
Administering these treatments shortly before or after the
symptomatic onset of epilepsy could effectively hinder the
advancement of spontaneous seizures and potentially delay their
onset (140). Moreover, it has
been demonstrated that IL-4 exerts a neuroprotective effect during
epileptogenesis by lowering TNF-α levels and mitigating
mitochondrial swelling in a mouse model induced by kaliotoxin
(141).
Mitochondrial dysfunction has a pivotal role in the
connection between epilepsy and oxidative stress (136). In total, ~40% of individuals
with epilepsy exhibit concomitant mitochondrial disorders (142). Mitochondrial dysfunction
disrupts the balance of RNS and ROS, resulting in heightened ROS
generation, oxidative harm and diminished ATP production. This
cascade ultimately culminates in mtDNA mutations and compromised
mitochondrial respiration, establishing a detrimental cycle
(1,136). Furthermore, there was a notable
rise in the occurrence of spontaneous motor seizures within
mitochondrial SOD2−/− mice (143). Conditional deletion of SOD2
specifically in the forebrain led to reduced mitochondrial oxygen
consumption and the subsequent development of epilepsy in mice
(42). An additional study
demonstrated that the targeted removal of neuron-specific
mitochondrial SOD2 results in a severe and intricate epileptic
phenotype (144). Collectively,
these findings indicate that mitochondrial oxidative stress is not
solely a result of epilepsy but also contributes to its onset.
Additionally, ROS can directly regulate
pro-inflammatory molecules, including IL-1β, high mobility group
box-1 (HMGB1) and matrix metalloproteinase 9 (136). Pathways related to HMGB1,
toll-like receptor 4 (TLR4) and IL-1β/interleukin-1 receptor 1 have
therefore emerged as potential targets for epilepsy therapy
(145). HMGB1 interacts with
TLR4 and functions as a proinflammatory cytokine in the
extracellular environment. Research has demonstrated that the
translocation of nuclear HMGB1 and active caspase-3 to the
mitochondria enhances programmed necrotic cell death in parvalbumin
cells and CA1 neurons during status epilepticus (146-148). Furthermore, another study
discovered that the levels of glutathione, an essential antioxidant
for maintaining mitochondrial integrity, significantly increased
following the administration of antioxidant drugs
[N-acetylcysteine (NAC) and sulforaphane]. Concurrently,
this intervention led to a reduction in HMGB1 production within an
acquired epilepsy rat model induced by status epilepticus (149,150). The NLRP3 inflammasome is a
molecule associated with epilepsy, which can be triggered by ROS
(151,152). Furthermore, it has been
empirically shown that mtROS function as a secondary messenger,
triggering the activation of NLRP3 and its translocation to the
mitochondria, thereby facilitating the activation of IL-1β.
Consequently, this process elicits a proinflammatory signal in
reaction to mitochondrial dysfunction (153,154). The available evidence suggests
that activation of the NLRP3 inflammasome can be inhibited by
anti-inflammatory and antioxidant therapy, thereby potentially
influencing epileptogenesis (155). A previous study also observed
upregulated levels of NLRP3 and IL-1β in children diagnosed with
febrile seizures (156). It has
also been suggested that NLRP1 and NLRP4 may have roles in the
development of epilepsy. Specifically, NLRP1 has been found to be
upregulated in patients with temporal lobe epilepsy (TLE) (157). In a TLE rat model, reducing the
expression of NLRP1 was shown to decrease the frequency and
severity of seizures (158).
The sensitivity of domain-containing protein 4 (NLRC4) to mtROS in
astrocytes and its association with mitochondrial oxidative stress
in neurodegenerative diseases provide insights into the potential
mechanism of NLRC4 in epilepsy (159). This highlights the intricate
relationship between epilepsy and mitochondrial dysfunction,
emphasizing the need to unravel the multifaceted impact of
mitochondrial function on brain activity as a potential avenue for
epilepsy treatment.
Mitochondrial therapy for neuroinflammation is an
emerging field with potential for the treatment of chronic
neuroinflammatory diseases (160). Currently, the overall treatment
strategy includes restoring normal mitochondrial physiological
functions (ATP production) and antioxidant therapy, clearing
mitochondria with abnormal functions through mitochondrial
autophagy or other mitochondrial stress responses, gene therapy,
restoring mitochondrial dynamics and addressing mitochondrial
calcium ion balance disorders (55,149,160-162).
Cytokines such as IL-1-β and TNF-α, as well as their
receptors, serve a significant role in the development and
progression of AD (55).
Inhibiting the interaction of these pro-inflammatory factors may
therefore be a more effective therapeutic option for AD. For
instance, the anti-inflammatory molecule, minocycline, reduces Aβ
and τ pathological lesions in an AD rat model by inhibiting
pro-inflammatory cytokines in glial cells through the NF-κB
signaling pathway (55).
Inhibiting inducible NO synthase and cyclooxygenase-2 is also
considered to effectively improve neuroinflammation in patients
with AD (163). Excessive
activation of microglial cells and reduced phagocytic ability leads
to increased accumulation of Aβ plaques and τ hyperphosphorylation,
exacerbating neuroinflammation (164). Microglial polarization from an
M1 state to a neuroprotective M2 state is also a potential target
for treatment. GV-971, a sodium oligomannate, modulates gut
microbiota amino acid metabolism to reduce the activation of T
helper 1 cells, thereby inhibiting M1-type microglia activation,
ultimately alleviating neuroinflammation and enhancing cognitive
function in AD mice (165). The
second-generation tetracycline, minocycline, selectively inhibits
the M1 state of microglial cells and exerts anti-neuroinflammatory
effects in patients with AD (166). Antioxidants are also
extensively researched in drug development. For instance, vitamin E
reduces the production of TNF-α and NO, lowering the levels of ROS
and IL-6 induced by lipopolysaccharides in microglial cells,
thereby providing neuroprotection (167). Polyphenolic compounds such as
flavonoids and vitamin C, may help prevent age-related
neurodegenerative diseases based on a clinical study (168). Flavonoids, which are found in
daily dietary products, promote the survival of neurons in patients
with AD by reducing protein oxidation, inhibiting the JNK and p38
pathways and preventing the production of free radicals (168).
Mitochondrial dysfunction is a well-established
feature of PD, with defects in mitochondrial complex I activity and
increased oxidative stress (169). In response to the
characteristics of this disease, the application of the
mitochondria-targeted antioxidant, mitoquinone (MitoQ), has been
gradually gaining attention. Cell experiments have confirmed that
MitoQ can reduce membrane leakage, oxidative stress and apoptosis
induced by α-synuclein (170).
In fruit flies with PINK1 knockout, vitamin K2, structurally
similar to coenzyme Q10 and also serving as an electron carrier in
the ETC, was found to alleviate oxidative stress in PD (162). However, in a double-blind
clinical study assessing untreated patients with PD using the
Unified Parkinson Disease Rating Scale, it was discovered that PD
did not improve after 12 months of MitoQ administration (171). Further research is therefore
needed to determine the effectiveness of MitoQ. Niacinamide
(Vitamin B3 and NAM) and its derivatives are currently under
investigation, with the aim to normalize redox levels (172). NAC has also been reported to
have demonstrated antioxidant properties in a clinical trial
(173). Additionally, a
promising candidate in recent clinical trials is ursodeoxycholic
acid (UDCA), known for its broad safety profile and its ability to
prevent mitochondrial membrane depolarization and stabilize
cytochrome c in mitochondria (174,175). The therapeutic potential of
UDCA in treating mitochondrial damage has been demonstrated in
LRRK2G2019S mutant PD patients and
LRRK2G2019S transgenic flies (161). Mitochondrial autophagy is also
a crucial target for PD treatment. In PD cell and mouse models,
celastrol plays a neuroprotective role by activating mitochondrial
autophagy and inhibiting DA neuron loss (176). Furthermore, mitochondrial
dynamics may represent a potential target for PD treatment. A study
has confirmed that the mitochondrial fission GTPase Drp1 inhibitor,
mdivi-1, can be used to inhibit mitochondrial fragmentation in
α-synuclein rat PD models, reducing neurodegeneration and
mitochondrial oxidative stress (177). Notably, it has been observed in
both animal models and patients with PD that physical exercise can
enhance mitochondrial biogenesis, providing new avenues for the
treatment of PD (178,179).
The treatment of MS is inherently complex due to
the varying subtypes of MS, each requiring distinct therapeutic
approaches. While significant progress has been made in the
treatment of MS, such as the effectiveness of the anti-CD20
antibody, ocrelizumab, and the sphingosine-1-phosphate receptor
(S1PR) modulator, siponimod, in patients with primary progressive
MS and relapsing-remitting MS (180,181), the management of other
progressive forms of MS remains challenging. For instance, in phase
III clinical trials, the S1PR modulator, fingolimod, did not
demonstrate a reduction in disability progression in patients with
primary progressive MS (182).
Immune-modulating compounds, such as siponimod and ocrelizumab,
targeting degenerative mechanisms may therefore not comprehensively
address neurodegenerative processes. Furthermore, the development
of new drugs is hindered by the incomplete understanding of the
pathogenesis of progressive MS and the absence of suitable animal
models. The onset of MS is often associated with the activation of
microglia and the continued involvement of T cells and B cells,
which release high levels of ROS and RNS, leading to mitochondrial
and axonal damage, and ultimately resulting in neurodegeneration.
Therefore, targeting mitochondria has emerged as a focal point in
the elucidation of methods to combat MS. Currently, mitochondrial
protective strategies, such as minocycline, iron (Fe2+)
chelating compounds and antioxidants that reduce oxidative stress,
have shown a certain degree of efficacy in MS treatment (119). Recent research has elucidated
that mitochondrial dysfunction impairs
Na+/K+-ATPase, leading to
Na+/Ca2+ exchanger reversal and calcium
overload, thereby mediating axonal degeneration (183). Notably, mitochondrial
transplantation into the medial forebrain bundle has been shown to
ameliorate motor deficits in 6-hydroxydopamine-induced PD rats,
enhancing mitochondrial functionality (184). It has also been demonstrated
that neural stem cells effectively deliver functional mitochondria
to target cells via extracellular vesicles, thereby remedying
mitochondrial functional deficits in mice with experimental
autoimmune encephalomyelitis (184). These studies therefore provide
evidence supporting the potential use of mitochondrial
transplantation as a therapeutic strategy for MS in the future.
Mitochondrial dysfunction is one of the most
prominent features of epilepsy, affecting 35-60% of patients with
epilepsy (185). Previous
research has found that cannabidiol (CBD) can reduce the frequency
of epileptic seizures (186).
This may be related to its ability to induce the formation of
mitochondrial-derived vesicles through the PINK1/Parkin pathway,
which participates in mitochondrial repair (187). Recent evidence suggests that
CBD engages in mitochondrial-related anti-inflammatory and
antioxidant activities, where it reverses iron-induced
mitochondrial dysfunction by rescuing mitochondrial ferritin and
modulating mtDNA epigenetics, and participates in neurodegenerative
mechanisms via the NF-κB, phosphorylated p38 MAPK and peroxisome
proliferation-activated receptor γ pathways (188). Currently, the Food and Drug
Administration has approved the drug compound, Epidiolex, which
contains CBD, for the treatment of seizures (189). Furthermore, a study has also
found that the IL-1 receptor antagonist, anakinra, can reduce
seizure frequency (190). The
antiepileptic drug, levetiracetam, can reduce neuronal excitability
by restoring the resting membrane potentials of IL-1β-induced
neurotoxic astrocytes and promoting the secretion of TGF-β1
(191). Additionally,
levetiracetam modulates the opening of the mPTP via synaptic
vesicle protein 2A, reducing neural hyperexcitability in patients
with AD and AD animal models (192). Other antioxidants targeting
mitochondria, such as polyphenols, vitamins and thiols, have been
shown to help reduce epileptic seizures (193). Therefore, targeting
mitochondria may be a key approach to treating epilepsy.
In general, the treatment strategies for chronic
neuroinflammatory diseases remain focused on combating oxidative
stress and ameliorating mitochondrial functional impairments, which
constitute shared pathophysiological features of such conditions.
Mitochondrially-targeted therapy stands out among emerging
therapeutic modalities due to its ability to selectively target
mitochondria, neutralizing reactive ROS and restoring their
functionality. Currently, research on agents such as MitoQ is the
most extensive. However, despite demonstrating significant
therapeutic effects in animal models, MitoQ has not yielded the
anticipated substantial benefits in clinical trials for PD or AD
(194,195). Moreover, effectively
penetrating the blood-brain barrier and achieving optimal
concentrations within target brain tissues remain pressing
challenges. Additionally, mitochondrial-targeted therapies fail to
selectively recognize damaged mitochondria and cannot directly
modulate mitochondrial dynamics and mitophagy processes, which may
contribute to their suboptimal clinical efficacy (194). In addition to
mitochondrial-targeted therapy, mitochondrial gene therapy is
emerging as a novel research domain. Treatment strategies encompass
restoring normal mitochondrial function, repairing or eliminating
mutated mtDNA and delivering wild-type mtDNA (196). Despite the development of
various delivery systems, including mitochondria-targeting peptides
and liposomes, as well as physical methods such as electroporation
and hydrodynamic injection, effectively delivering therapeutic
macromolecules to mitochondria remains a challenge due to the
presence of the blood-brain barrier (196). Furthermore, delivery systems
may induce cytotoxicity or interact with endogenous biomolecules,
leading to aggregation and reduced efficacy (197). The development of mitochondrial
genome editing technology is still in its nascent stages,
necessitating further understanding of how RNA and editing tools
penetrate mammalian mitochondria (198). Efforts to develop more precise
and safer mitochondrial-targeted drugs may therefore be a future
research focus.
Mitochondrial dysfunction is a common feature of
chronic neuroinflammatory diseases and exploring its pathological
mechanisms may provide new avenues for future treatments. Various
drugs have been developed that target mitochondria, focusing on
aspects such as antioxidation, mitochondrial autophagy regulation,
calcium ion balance and gene repair. However, clinical application
of these drugs remains a significant challenge. Exploring new
therapeutic targets, selectively targeting dysfunctional
mitochondria, ensuring delivery of drugs across the blood-brain
barrier into the brain and minimizing adverse reactions may be the
focus of future research. Additionally, advancements in
mitochondrial genome editing technology offer hope for the precise
manipulation of mitochondrial function and addressing genetic
abnormalities in neuroinflammatory diseases.
Future treatment strategies may not be limited to a
single approach; combining anti-inflammatory and antioxidative
therapy with mitochondrial-targeted treatment may enhance overall
treatment safety and efficacy. In conclusion, while
mitochondrial-targeted therapy holds promise for the treatment of
chronic neuroinflammatory diseases, addressing current limitations
is crucial. By overcoming delivery challenges, enhancing treatment
specificity and exploring new therapeutic targets,
mitochondrial-targeted therapy holds promise for treating chronic
neuroinflammatory diseases and other neurological disorders.
Not applicable.
PQ and YS contributed equally to this work.
Conceptualization, writing the original draft and reviewing and
editing the manuscript was conducted by LL; writing the original
draft and reviewing the manuscript was conducted by PQ; Reviewing
and editing the manuscript and drawing the figures was conducted by
YS. All authors read and approved the final version of the
manuscript. Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
No funding was received.
|
1
|
Vezzani B, Carinci M, Patergnani S,
Pasquin MP, Guarino A, Aziz N, Pinton P, Simonato M and Giorgi C:
The Dichotomous role of inflammation in the CNS: A mitochondrial
point of view. Biomolecules. 10:14372020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yilmaz C, Karali K, Fodelianaki G,
Gravanis A, Chavakis T, Charalampopoulos I and Alexaki VI:
Neurosteroids as regulators of neuroinflammation. Front
Neuroendocrinol. 55:1007882019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fontana L, Ghezzi L, Cross AH and Piccio
L: Effects of dietary restriction on neuroinflammation in
neurodegenerative diseases. J Exp Med. 218:e201900862021.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Biswas K: Microglia mediated
neuroinflammation in neurodegenerative diseases: A review on the
cell signaling pathways involved in microglial activation. J
Neuroimmunol. 383:5781802023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu A, Yu L, Li X, Zhang K, Zhang W, So
KF, Tissir F, Qu Y and Zhou L: Celsr2-mediated morphological
polarization and functional phenotype of reactive astrocytes in
neural repair. Glia. 71:1985–2004. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fan YY and Huo J: A1/A2 astrocytes in
central nervous system injuries and diseases: Angels or devils?
Neurochem Int. 148:1050802021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Singh D: Astrocytic and microglial cells
as the modulators of neuroinflammation in Alzheimer's disease. J
Neuroinflammation. 19:2062022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gimenez MA, Sim J, Archambault AS, Klein
RS and Russell JH: A tumor necrosis factor receptor 1-dependent
conversation between central nervous system-specific T cells and
the central nervous system is required for inflammatory
infiltration of the spinal cord. Am J Pathol. 168:1200–1209. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Da Mesquita S, Fu Z and Kipnis J: The
meningeal lymphatic system: A new player in neurophysiology.
Neuron. 100:375–388. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Flores-Romero H, Dadsena S and Garcia-Saez
AJ: Mitochondrial pores at the crossroad between cell death and
inflammatory signaling. Mol Cell. 83:843–856. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wei Y, Miao Q, Zhang Q, Mao S, Li M, Xu X,
Xia X, Wei K, Fan Y, Zheng X, et al: Aerobic glycolysis is the
predominant means of glucose metabolism in neuronal somata, which
protects against oxidative damage. Nat Neurosci. 26:2081–2089.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rose J, Brian C, Woods J, Pappa A,
Panayiotidis MI, Powers R and Franco R: Mitochondrial dysfunction
in glial cells: Implications for neuronal homeostasis and survival.
Toxicology. 391:109–115. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rizzuto R, De Stefani D, Raffaello A and
Mammucari C: Mitochondria as sensors and regulators of calcium
signalling. Nat Rev Mol Cell Biol. 13:566–578. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Satarker S, Bojja SL, Gurram PC, Mudgal J,
Arora D and Nampoothiri M: Astrocytic glutamatergic transmission
and its implications in neurodegenerative disorders. Cells.
11:11392022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Morales-Ropero JM, Arroyo-Urea S, Neubrand
VE, Martín-Oliva D, Marín-Teva JL, Cuadros MA, Vangheluwe P,
Navascués J, Mata AM and Sepúlveda MR: The endoplasmic reticulum
Ca(2+) -ATPase SERCA2b is upregulated in activated microglia and
its inhibition causes opposite effects on migration and
phagocytosis. Glia. 69:842–857. 2021. View Article : Google Scholar
|
|
16
|
Neel DV, Basu H, Gunner G, Bergstresser
MD, Giadone RM, Chung H, Miao R, Chou V, Brody E, Jiang X, et al:
Gasdermin-E mediates mitochondrial damage in axons and
neurodegeneration. Neuron. 111:1222–1240 e1229. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Borsche M, Pereira SL, Klein C and
Grünewald A: Mitochondria and Parkinson's disease: Clinical,
molecular, and translational aspects. J Parkinsons Dis. 11:45–60.
2021. View Article : Google Scholar :
|
|
18
|
Hinkle JT, Patel J, Panicker N,
Karuppagounder SS, Biswas D, Belingon B, Chen R, Brahmachari S,
Pletnikova O, Troncoso JC, et al: STING mediates neurodegeneration
and neuroinflammation in nigrostriatal α-synucleinopathy. Proc Natl
Acad Sci USA. 119:e21188191192022. View Article : Google Scholar
|
|
19
|
Pezone A, Olivieri F, Napoli MV, Procopio
A, Avvedimento EV and Gabrielli A: inflammation and DNA damage:
Cause, effect or both. Nat Rev Rheumatol. 19:200–211. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Duarte JN: Neuroinflammatory mechanisms of
mitochondrial dysfunction and neurodegeneration in glaucoma. J
Ophthalmol. 2021:45819092021.PubMed/NCBI
|
|
21
|
Yang Y, Liu Y, Zhu J, Song S, Huang Y,
Zhang W, Sun Y, Hao J, Yang X, Gao Q, et al:
Neuroinflammation-mediated mitochondrial dysregulation involved in
postoperative cognitive dysfunction. Free Radic Biol Med.
178:134–146. 2022. View Article : Google Scholar
|
|
22
|
Pan RY, Ma J, Kong XX, Wang XF, Li SS, Qi
XL, Yan YH, Cheng J, Liu Q, Jin W, et al: Sodium rutin ameliorates
Alzheimer's disease-like pathology by enhancing microglial
amyloid-β clearance. Sci Adv. 5:eaau63282019. View Article : Google Scholar
|
|
23
|
Joshi AU, Minhas PS, Liddelow SA,
Haileselassie B, Andreasson KI, Dorn GW II and Mochly-Rosen D:
Fragmented mitochondria released from microglia trigger A1
astrocytic response and propagate inflammatory neurodegeneration.
Nat Neurosci. 22:1635–1648. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mi Y, Qi G, Vitali F, Shang Y, Raikes AC,
Wang T, Jin Y, Brinton RD, Gu H and Yin F: Loss of fatty acid
degradation by astrocytic mitochondria triggers neuroinflammation
and neurodegeneration. Nat Metab. 5:445–465. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cenini G, Rub C, Bruderek M and Voos W:
Amyloid β-peptides interfere with mitochondrial preprotein import
competence by a coaggregation process. Mol Biol Cell. 27:3257–3272.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bingol B, Tea JS, Phu L, Reichelt M,
Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS and Sheng M: The
mitochondrial deubiquitinase USP30 opposes parkin-mediated
mitophagy. Nature. 510:370–375. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Annesley SJ and Fisher PR: Mitochondria in
health and disease. Cells. 8:6802019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lin MT and Beal MF: Mitochondrial
dysfunction and oxidative stress in neurodegenerative diseases.
Nature. 443:787–795. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Johnson J, Mercado-Ayon E, Mercado-Ayon Y,
Dong YN, Halawani S, Ngaba L and Lynch DR: Mitochondrial
dysfunction in the development and progression of neurodegenerative
diseases. Arch Biochem Biophys. 702:1086982021. View Article : Google Scholar
|
|
30
|
Adebayo M, Singh S, Singh AP and Dasgupta
S: Mitochondrial fusion and fission: The fine-tune balance for
cellular homeostasis. FASEB J. 35:e216202021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Alevriadou BR, Patel A, Noble M, Ghosh S,
Gohil VM, Stathopulos PB and Madesh M: Molecular nature and
physiological role of the mitochondrial calcium uniporter channel.
Am J Physiol Cell Physiol. 320:C465–C482. 2021. View Article : Google Scholar :
|
|
32
|
Fischer R and Maier O: Interrelation of
oxidative stress and inflammation in neurodegenerative disease:
Role of TNF. Oxid Med Cell Longev. 2015:6108132015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Teleanu DM, Niculescu AG, Lungu II, Radu
CI, Vladâcenco O, Roza E, Costăchescu B, Grumezescu AM and Teleanu
RI: An overview of oxidative stress, neuroinflammation, and
neurodegenerative diseases. Int J Mol Sci. 23:59382022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tsang T, Davis CI and Brady DC: Copper
biology. Curr Biol. 31:R421–R427. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Borisov VB, Siletsky SA, Nastasi MR and
Forte E: ROS defense systems and terminal oxidases in bacteria.
Antioxidants (Basel). 10:8392021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Haider S, Batool Z, Ahmad S, Siddiqui RA
and Haleem DJ: Walnut supplementation reverses the
scopolamine-induced memory impairment by restoration of cholinergic
function via mitigating oxidative stress in rats: A potential
therapeutic intervention for age related neurodegenerative
disorders. Metab Brain Dis. 33:39–51. 2018. View Article : Google Scholar
|
|
37
|
Luque-Contreras D, Carvajal K, Toral-Rios
D, Franco-Bocanegra D and Campos-Pena V: Oxidative stress and
metabolic syndrome: Cause or consequence of Alzheimer's disease?
Oxid Med Cell Longev. 2014:4978022014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Singh A, Kukreti R, Saso L and Kukreti S:
Oxidative stress: A key modulator in neurodegenerative diseases.
Molecules. 24:15832019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bhatia V and Sharma S: Role of
mitochondrial dysfunction, oxidative stress and autophagy in
progression of Alzheimer's disease. J Neurol Sci. 421:1172532021.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS
release. Physiol Rev. 94:909–950. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Farkhondeh T, Mehrpour O, Forouzanfar F,
Roshanravan B and Samarghandian S: Oxidative stress and
mitochondrial dysfunction in organophosphate pesticide-induced
neurotoxicity and its amelioration: A review. Environ Sci Pollut
Res Int. 27:24799–24814. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rowley S, Liang LP, Fulton R, Shimizu T,
Day B and Patel M: Mitochondrial respiration deficits driven by
reactive oxygen species in experimental temporal lobe epilepsy.
Neurobiol Dis. 75:151–158. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bordoni L and Gabbianelli R: Mitochondrial
DNA and neurodegeneration: Any role for dietary antioxidants?
Antioxidants (Basel). 9:7642020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Coppede F: Mitochondrial DNA methylation
and mitochondria-related epigenetics in neurodegeneration. Neural
Regen Res. 19:405–406. 2024. View Article : Google Scholar
|
|
45
|
Sharma VK, Singh TG and Mehta V: Stressed
mitochondria: A target to intrude alzheimer's disease.
Mitochondrion. 59:48–57. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lin J and Epel E: Stress and telomere
shortening: Insights from cellular mechanisms. Ageing Res Rev.
73:1015072022. View Article : Google Scholar :
|
|
47
|
Ortiz JM and Swerdlow RH: Mitochondrial
dysfunction in Alzheimer's disease: Role in pathogenesis and novel
therapeutic opportunities. Br J Pharmacol. 176:3489–3507. 2019.
View Article : Google Scholar
|
|
48
|
Liu H, Zhang H, Zhang Y, Xu S, Zhao H, He
H and Liu X: Modeling mtDNA hypermethylation vicious circle
mediating Aβ-induced endothelial damage memory in HCMEC/D3 cell.
Aging (Albany NY). 12:18343–18362. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Rasheed M, Liang J, Wang C, Deng Y and
Chen Z: Epigenetic regulation of neuroinflammation in Parkinson's
disease. Int J Mol Sci. 22:49562021. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Youle RJ and van der Bliek AM:
Mitochondrial fission, fusion, and stress. Science. 337:1062–1065.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Song Y, Xu Y, Liu Y, Gao J, Feng L, Zhang
Y, Shi L and Zhang M, Guo D, Qi B and Zhang M: Mitochondrial
quality control in the maintenance of cardiovascular homeostasis:
The roles and interregulation of UPS, mitochondrial dynamics and
mitophagy. Oxid Med Cell Longev. 2021:39607732021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Xie JH, Li YY and Jin J: The essential
functions of mitochondrial dynamics in immune cells. Cell Mol
Immunol. 17:712–721. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chakrabarti S and Bisaglia M: Oxidative
stress and neuroinflammation in Parkinson's disease: The role of
dopamine oxidation products. Antioxidants (Basel). 12:9552023.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Picca A, Ferri E, Calvani R, Coelho-Junior
HJ, Marzetti E and Arosio B: Age-Associated glia remodeling and
mitochondrial dysfunction in neurodegeneration: Antioxidant
supplementation as a possible intervention. Nutrients. 14:24062022.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Dhapola R, Hota SS, Sarma P, Bhattacharyya
A, Medhi B and Reddy DH: Recent advances in molecular pathways and
therapeutic implications targeting neuroinflammation for
Alzheimer's disease. Inflammopharmacology. 29:1669–1681. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chang YH, Lin HY, Shen FC, Su YJ, Chuang
JH, Lin TK, Liou CW, Lin CY, Weng SW and Wang PW: The causal role
of mitochondrial dynamics in regulating innate immunity in
diabetes. Front Endocrinol (Lausanne). 11:4452020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Paik S, Kim JK, Silwal P, Sasakawa C and
Jo EK: An update on the regulatory mechanisms of NLRP3 inflammasome
activation. Cell Mol Immunol. 18:1141–1160. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yurtsever I, Ustundag UV, Ünal I, Ateş PS
and Emekli-Alturfan E: Rifampicin decreases neuroinflammation to
maintain mitochondrial function and calcium homeostasis in
rotenone-treated zebrafish. Drug Chem Toxicol. 45:1544–1551. 2022.
View Article : Google Scholar
|
|
59
|
Kannurpatti SS: Mitochondrial calcium
homeostasis: Implications for neurovascular and neurometabolic
coupling. J Cereb Blood Flow Metab. 37:381–395. 2017. View Article : Google Scholar :
|
|
60
|
Poewe W, Seppi K, Tanner CM, Halliday GM,
Brundin P, Volkmann J, Schrag AE and Lang AE: Parkinson disease.
Nat Rev Dis Primers. 3:170132017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Binvignat O and Olloquequi J:
Excitotoxicity as a target against neurodegenerative processes.
Curr Pharm Des. 26:1251–1262. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Yao L, Wu J, Koc S and Lu G: Genetic
imaging of neuroinflammation in Parkinson's disease: Recent
advancements. Front Cell Dev Biol. 9:6558192021. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Obrador E, Salvador R, Lopez-Blanch R,
Jihad-Jebbar A, Valles SL and Estrela JM: Oxidative stress,
neuroinflammation and mitochondria in the pathophysiology of
amyotrophic lateral sclerosis. Antioxidants (Basel). 9:9012020.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Cyrino LAR, Delwing-de Lima D, Ullmann OM
and Maia TP: Concepts of neuroinflammation and their relationship
with impaired mitochondrial functions in bipolar disorder. Front
Behav Neurosci. 15:6094872021. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Verkhratsky A, Rodriguez-Arellano JJ,
Parpura V and Zorec R: Astroglial calcium signalling in Alzheimer's
disease. Biochem Biophys Res Commun. 483:1005–1012. 2017.
View Article : Google Scholar
|
|
66
|
Casaril AM, Katsalifis A, Schmidt RM and
Bas-Orth C: Activated glia cells cause bioenergetic impairment of
neurons that can be rescued by knock-down of the mitochondrial
calcium uniporter. Biochem Biophys Res Commun. 608:45–51. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Garbincius JF and Elrod JW: Mitochondrial
calcium exchange in physiology and disease. Physiol Rev.
102:893–992. 2022. View Article : Google Scholar :
|
|
68
|
Baumgartner HK, Gerasimenko JV, Thorne C,
Ferdek P, Pozzan T, Tepikin AV, Petersen OH, Sutton R, Watson AJ
and Gerasimenko OV: Calcium elevation in mitochondria is the main
Ca2+ requirement for mitochondrial permeability transition pore
(mPTP) opening. J Biol Chem. 284:20796–20803. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Green DR and Kroemer G: The
pathophysiology of mitochondrial cell death. Science. 305:626–629.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Rimessi A, Previati M, Nigro F, Wieckowski
MR and Pinton P: Mitochondrial reactive oxygen species and
inflammation: Molecular mechanisms, diseases and promising
therapies. Int J Biochem Cell Biol. 81:281–293. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Marchi S, Patergnani S, Missiroli S,
Morciano G, Rimessi A, Wieckowski MR, Giorgi C and Pinton P:
Mitochondrial and endoplasmic reticulum calcium homeostasis and
cell death. Cell Calcium. 69:62–72. 2018. View Article : Google Scholar
|
|
72
|
Ooi K, Hu L, Feng Y, Han C, Ren X, Qian X,
Huang H, Chen S, Shi Q, Lin H, et al: Sigma-1 receptor activation
suppresses microglia M1 polarization via regulating endoplasmic
reticulum-mitochondria contact and mitochondrial functions in
stress-induced hypertension rats. Mol Neurobiol. 58:6625–6646.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Harland M, Torres S, Liu J and Wang X:
Neuronal mitochondria modulation of LPS-induced neuroinflammation.
J Neurosci. 40:1756–1765. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Lim D, Dematteis G, Tapella L, Genazzani
AA, Calì T, Brini M and Verkhratsky A: Ca(2+) handling at the
mitochondria-ER contact sites in neurodegeneration. Cell Calcium.
98:1024532021. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Krols M, van Isterdael G, Asselbergh B,
Kremer A, Lippens S, Timmerman V and Janssens S:
Mitochondria-associated membranes as hubs for neurodegeneration.
Acta Neuropathol. 131:505–523. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Sunanda T, Ray B, Mahalakshmi AM, Bhat A,
Rashan L, Rungratanawanich W, Song BJ, Essa MM, Sakharkar MK and
Chidambaram SB: Mitochondria-endoplasmic reticulum crosstalk in
Parkinson's disease: The role of brain renin angiotensin system
components. Biomolecules. 11:16692021. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Du H, Guo L, Fang F, Chen D, Sosunov AA,
McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, et al:
Cyclophilin D deficiency attenuates mitochondrial and neuronal
perturbation and ameliorates learning and memory in Alzheimer's
disease. Nat Med. 14:1097–1105. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Ayabe T, Takahashi C, Ohya R and Ano Y:
β-Lactolin improves mitochondrial function in Abeta-treated mouse
hippocampal neuronal cell line and a human iPSC-derived neuronal
cell model of Alzheimer's disease. FASEB J. 36:e222772022.
View Article : Google Scholar
|
|
79
|
Reddy PH and Beal MF: Amyloid beta,
mitochondrial dysfunction and synaptic damage: Implications for
cognitive decline in aging and Alzheimer's disease. Trends Mol Med.
14:45–53. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Dursun E, Alaylioglu M, Bilgic B, Hanağası
H, Gürvit H, Emre M and Gezen-Ak D: Amyloid beta adsorption problem
with transfer plates in amyloid beta 1-42 IVD Kits. J Mol Neurosci.
67:534–539. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Han J, Park H, Maharana C, Gwon AR, Park
J, Baek SH, Bae HG, Cho Y, Kim HK, Sul JH, et al: Alzheimer's
disease-causing presenilin-1 mutations have deleterious effects on
mitochondrial function. Theranostics. 11:8855–8873. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Bai R, Guo J, Ye XY, Xie Y and Xie T:
Oxidative stress: The core pathogenesis and mechanism of
Alzheimer's disease. Ageing Res Rev. 77:1016192022. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Kowalczyk P, Sulejczak D, Kleczkowska P,
Bukowska-Ośko I, Kucia M, Popiel M, Wietrak E, Kramkowski K,
Wrzosek K and Kaczyńska K: Mitochondrial oxidative stress-A
causative factor and therapeutic target in many diseases. Int J Mol
Sci. 22:133842021. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Park MW, Cha HW, Kim J, Kim JH, Yang H,
Yoon S, Boonpraman N, Yi SS, Yoo ID and Moon JS: NOX4 promotes
ferroptosis of astrocytes by oxidative stress-induced lipid
peroxidation via the impairment of mitochondrial metabolism in
Alzheimer's diseases. Redox Biol. 41:1019472021. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Islam MT: Oxidative stress and
mitochondrial dysfunction-linked neurodegenerative disorders.
Neurol Res. 39:73–82. 2017. View Article : Google Scholar
|
|
86
|
Simpson DSA and Oliver PL: ROS generation
in microglia: Understanding oxidative stress and inflammation in
neurodegenerative disease. Antioxidants (Basel). 9:7432020.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
ElAli A, Bordeleau M, Theriault P, Filali
M, Lampron A and Rivest S: Tissue-plasminogen activator attenuates
Alzheimer's disease-related pathology development in APPswe/PS1
mice. Neuropsychopharmacology. 41:1297–1307. 2016. View Article : Google Scholar :
|
|
88
|
Li Y, Xia X, Wang Y and Zheng JC:
Mitochondrial dysfunction in microglia: A novel perspective for
pathogenesis of Alzheimer's disease. J Neuroinflammation.
19:2482022. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Massey N, Shrestha D, Bhat SM, Kondru N,
Charli A, Karriker LA, Kanthasamy AG and Charavaryamath C: Organic
dust-induced mitochondrial dysfunction could be targeted via
cGAS-STING or cytoplasmic NOX-2 inhibition using microglial cells
and brain slice culture models. Cell Tissue Res. 384:465–486. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Ising C, Venegas C, Zhang S, Scheiblich H,
Schmidt SV, Vieira-Saecker A, Schwartz S, Albasset S, McManus RM,
Tejera D, et al: NLRP3 inflammasome activation drives tau
pathology. Nature. 575:669–673. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Kelley N, Jeltema D, Duan Y and He Y: The
NLRP3 inflammasome: An overview of mechanisms of activation and
regulation. Int J Mol Sci. 20:33282019. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Jassim AH, Inman DM and Mitchell CH:
Crosstalk between dysfunctional mitochondria and inflammation in
glaucomatous neurodegeneration. Front Pharmacol. 12:6996232021.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Shaftel SS, Griffin WS and O'Banion MK:
The role of interleukin-1 in neuroinflammation and Alzheimer
disease: An evolving perspective. J Neuroinflammation. 5:72008.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Gonzalez-Reyes RE, Nava-Mesa MO,
Vargas-Sanchez K, Ariza-Salamanca D and Mora-Munoz L: Involvement
of astrocytes in Alzheimer's disease from a neuroinflammatory and
oxidative stress perspective. Front Mol Neurosci. 10:4272017.
View Article : Google Scholar
|
|
95
|
Sheng JG, Ito K, Skinner RD, Mrak RE,
Rovnaghi CR, Van Eldik LJ and Griffin WS: In vivo and in vitro
evidence supporting a role for the inflammatory cytokine
interleukin-1 as a driving force in Alzheimer pathogenesis.
Neurobiol Aging. 17:761–766. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Bossu P, Ciaramella A, Moro ML,
Bellincampi L, Bernardini S, Federici G, Trequattrini A, Macciardi
F, Spoletini I, Di Iulio F, et al: Interleukin 18 gene
polymorphisms predict risk and outcome of Alzheimer's disease. J
Neurol Neurosurg Psychiatry. 78:807–811. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Gasser T: Molecular pathogenesis of
Parkinson disease: Insights from genetic studies. Expert Rev Mol
Med. 11:e222009. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Braak H, Del Tredici K, Rub U, de Vos RA,
Jansen EN and Braak E: Staging of brain pathology related to
sporadic Parkinson's disease. Neurobiol Aging. 24:197–211. 2003.
View Article : Google Scholar
|
|
99
|
Han C, Liu Y, Dai R, Ismail N, Su W and Li
B: Ferroptosis and its potential role in human diseases. Front
Pharmacol. 11:2392020. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Panicker N, Ge P, Dawson VL and Dawson TM:
The cell biology of Parkinson's disease. J Cell Biol.
220:e2020120952021. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Liu G, Zhang C, Yin J, Li X, Cheng F, Li
Y, Yang H, Uéda K, Chan P and Yu S: alpha-Synuclein is
differentially expressed in mitochondria from different rat brain
regions and dose-dependently down-regulates complex I activity.
Neurosci Lett. 454:187–192. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Thomas RR, Keeney PM and Bennett JP:
Impaired complex-I mitochondrial biogenesis in Parkinson disease
frontal cortex. J Parkinsons Dis. 2:67–76. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Bender A, Krishnan KJ, Morris CM, Taylor
GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock
T, et al: High levels of mitochondrial DNA deletions in substantia
nigra neurons in aging and Parkinson disease. Nat Genet.
38:515–517. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
104
|
Valente EM, Bentivoglio AR, Dixon PH,
Ferraris A, Ialongo T, Frontali M, Albanese A and Wood NW:
Localization of a novel locus for autosomal recessive early-onset
parkinsonism, PARK6, on human chromosome 1p35-p36. Am J Hum Genet.
68:895–900. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
105
|
Fan Z, Pan YT, Zhang ZY, Yang H, Yu SY,
Zheng Y, Ma JH and Wang XM: Systemic activation of NLRP3
inflammasome and plasma α-synuclein levels are correlated with
motor severity and progression in Parkinson's disease. J
Neuroinflammation. 17:112020. View Article : Google Scholar
|
|
106
|
Sliter DA, Martinez J, Hao L, Chen X, Sun
N, Fischer TD, Burman JL, Li Y, Zhang Z and Narendra DP: Parkin and
PINK1 mitigate STING-induced inflammation. Nature. 561:258–262.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Hou X, Fiesel FC, Truban D, Casey MC, Lin
WL, Soto AI, Tacik P, Rousseau LG, Diehl NN, Heckman MG, et al:
Age- and disease-dependent increase of the mitophagy marker
phospho-ubiquitin in normal aging and Lewy body disease. Autophagy.
14:1404–1418. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Di Maio R, Barrett PJ, Hoffman EK, Barrett
CW, Zharikov A, Borah A, Hu X, McCoy J, Chu CT, Burton EA, et al:
α-synuclein binds to TOM20 and inhibits mitochondrial protein
import in Parkinson's disease. Sci Transl Med. 8:342ra3782016.
View Article : Google Scholar
|
|
109
|
Vicario M, Cieri D, Vallese F, Catoni C,
Barazzuol L, Berto P, Grinzato A, Barbieri L, Brini M and Calì T: A
split-GFP tool reveals differences in the sub-mitochondrial
distribution of wt and mutant alpha-synuclein. Cell Death Dis.
10:8572019. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Devi L, Raghavendran V, Prabhu BM,
Avadhani NG and Anandatheerthavarada HK: Mitochondrial import and
accumulation of alpha-synuclein impair complex I in human
dopaminergic neuronal cultures and Parkinson disease brain. J Biol
Chem. 283:9089–9100. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Mahul-Mellier AL, Burtscher J, Maharjan N,
Weerens L, Croisier M, Kuttler F, Leleu M, Knott GW and Lashuel HA:
The process of Lewy body formation, rather than simply
alpha-synuclein fibrillization, is one of the major drivers of
neurodegeneration. Proc Natl Acad Sci USA. 117:4971–4982. 2020.
View Article : Google Scholar
|
|
112
|
Hsieh CH, Shaltouki A, Gonzalez AE, da
Cruz AB, Burbulla LF, St Lawrence E, Schüle B, Krainc D, Palmer TD
and Wang X: Functional impairment in miro degradation and mitophagy
is a shared feature in familial and sporadic Parkinson's disease.
Cell Stem Cell. 19:709–724. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Godena VK, Brookes-Hocking N, Moller A,
Shaw G, Oswald M, Sancho RM, Miller CC, Whitworth AJ and De Vos KJ:
Increasing microtubule acetylation rescues axonal transport and
locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat
Commun. 5:52452014. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Bonello F, Hassoun SM, Mouton-Liger F,
Shin YS, Muscat A, Tesson C, Lesage S, Bear PM, Brice A, Krupp J,
et al: LRRK2 impairs PINK1/Parkin-dependent mitophagy via its
kinase activity: Pathologic insights into Parkinson's disease. Hum
Mol Genet. 28:1645–1660. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Wang X, Chi J, Huang D, Ding L, Zhao X,
Jiang L, Yu Y and Gao F: α-synuclein promotes progression of
Parkinson's disease by upregulating autophagy signaling pathway to
activate NLRP3 inflammasome. Exp Ther Med. 19:931–938.
2020.PubMed/NCBI
|
|
116
|
Sarkar S, Malovic E, Harishchandra DS,
Ghaisas S, Panicker N, Charli A, Palanisamy BN, Rokad D, Jin H,
Anantharam V, et al: Mitochondrial impairment in microglia
amplifies NLRP3 inflammasome proinflammatory signaling in cell
culture and animal models of Parkinson's disease. NPJ Parkinsons
Dis. 3:302017. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Mouton-Liger F, Rosazza T, Sepulveda-Diaz
J, Ieang A, Hassoun SM, Claire E, Mangone G, Brice A, Michel PP,
Corvol JC and Corti O: Parkin deficiency modulates NLRP3
inflammasome activation by attenuating an A20-dependent negative
feedback loop. Glia. 66:1736–1751. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Zhang C, Zhao M, Wang B, Su Z, Guo B, Qin
L, Zhang W and Zheng R: The Nrf2-NLRP3-caspase-1 axis mediates the
neuroprotective effects of celastrol in Parkinson's disease. Redox
Biol. 47:1021342021. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Faissner S, Plemel JR, Gold R and Yong VW:
Progressive multiple sclerosis: From pathophysiology to therapeutic
strategies. Nat Rev Drug Discov. 18:905–922. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Barcelos IP, Troxell RM and Graves JS:
Mitochondrial dysfunction and multiple sclerosis. Biology (Basel).
8:372019.PubMed/NCBI
|
|
121
|
Steinman L: Multiple sclerosis: A
two-stage disease. Nat Immunol. 2:762–764. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Lassmann H, van Horssen J and Mahad D:
Progressive multiple sclerosis: Pathology and pathogenesis. Nat Rev
Neurol. 8:647–656. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Lopez-Domenech G and Kittler JT:
Mitochondrial regulation of local supply of energy in neurons. Curr
Opin Neurobiol. 81:1027472023. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Touil H, Li R, Zuroff L, Moore CS, Healy
L, Cignarella F, Piccio L, Ludwin S, Prat A, Gommerman J, et al:
Cross-talk between B cells, microglia and macrophages, and
implications to central nervous system compartmentalized
inflammation and progressive multiple sclerosis. EBioMedicine.
96:1047892023. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Campbell GR, Ziabreva I, Reeve AK,
Krishnan KJ, Reynolds R, Howell O, Lassmann H, Turnbull DM and
Mahad DJ: Mitochondrial DNA deletions and neurodegeneration in
multiple sclerosis. Ann Neurol. 69:481–492. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Witte ME, Nijland PG, Drexhage JA,
Gerritsen W, Geerts D, van Het Hof B, Reijerkerk A, de Vries HE,
van der Valk P and van Horssen J: Reduced expression of PGC-1alpha
partly underlies mitochondrial changes and correlates with neuronal
loss in multiple sclerosis cortex. Acta Neuropathol. 125:231–243.
2013. View Article : Google Scholar
|
|
127
|
Haider L, Fischer MT, Frischer JM, Bauer
J, Höftberger R, Botond G, Esterbauer H, Binder CJ, Witztum JL and
Lassmann H: Oxidative damage in multiple sclerosis lesions. Brain.
134:1914–1924. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Dziedzic T, Metz I, Dallenga T, König FB,
Müller S, Stadelmann C and Brück W: Wallerian degeneration: A major
component of early axonal pathology in multiple sclerosis. Brain
Pathol. 20:976–985. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Madsen PM, Pinto M, Patel S, McCarthy S,
Gao H, Taherian M, Karmally S, Pereira CV, Dvoriantchikova G,
Ivanov D, et al: Mitochondrial DNA double-strand breaks in
oligodendrocytes cause demyelination, axonal injury, and CNS
inflammation. J Neurosci. 37:10185–10199. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Singhal NK, Alkhayer K, Shelestak J,
Clements R, Freeman E and McDonough J: Erythropoietin upregulates
brain hemoglobin expression and supports neuronal mitochondrial
activity. Mol Neurobiol. 55:8051–8058. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Aboul-Enein F, Krssak M, Hoftberger R,
Prayer D and Kristoferitsch W: Reduced NAA-levels in the NAWM of
patients with MS is a feature of progression. A study with
quantitative magnetic resonance spectroscopy at 3 tesla. PLoS One.
5:e116252010. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Dominicis A, Del Giovane A, Torreggiani M,
Recchia AD, Ciccarone F, Ciriolo MR and Ragnini-Wilson A:
N-Acetylaspartate drives oligodendroglial differentiation via
histone deacetylase activation. Cells. 12:18612023. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Kadowaki A and Quintana FJ: The NLRP3
inflammasome in progressive multiple sclerosis. Brain.
143:1286–1288. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Ferecsko AS, Smallwood MJ, Moore A, Liddle
C, Newcombe J, Holley J, Whatmore J, Gutowski NJ and Eggleton P:
STING-triggered CNS inflammation in human neurodegenerative
diseases. Biomedicines. 11:13752023. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Thijs RD, Surges R, O'Brien TJ and Sander
JW: Epilepsy in adults. Lancet. 393:689–701. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Fabisiak T and Patel M: Crosstalk between
neuroinflammation and oxidative stress in epilepsy. Front Cell Dev
Biol. 10:9769532022. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Loscher W and Klein P: The pharmacology
and clinical efficacy of antiseizure medications: From bromide
salts to cenobamate and beyond. CNS Drugs. 35:935–963. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Pearson-Smith JN and Patel M: Metabolic
dysfunction and oxidative stress in epilepsy. Int J Mol Sci.
18:23652017. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Geronzi U, Lotti F and Grosso S: Oxidative
stress in epilepsy. Expert Rev Neurother. 18:427–434. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Terrone G, Balosso S, Pauletti A, Ravizza
T and Vezzani A: Inflammation and reactive oxygen species as
disease modifiers in epilepsy. Neuropharmacology. 167:1077422020.
View Article : Google Scholar
|
|
141
|
Ahras-Sifi N and Laraba-Djebari F:
Immunomodulatory and protective effects of interleukin-4 on the
neuropathological alterations induced by a potassium channel
blocker. J Neuroimmunol. 355:5775492021. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Rahman S: Pathophysiology of mitochondrial
disease causing epilepsy and status epilepticus. Epilepsy Behav.
49:71–75. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Liang LP, Waldbaum S, Rowley S, Huang TT,
Day BJ and Patel M: Mitochondrial oxidative stress and epilepsy in
SOD2 deficient mice: Attenuation by a lipophilic metalloporphyrin.
Neurobiol Dis. 45:1068–1076. 2012. View Article : Google Scholar :
|
|
144
|
Fulton RE, Pearson-Smith JN, Huynh CQ,
Fabisiak T, Liang LP, Aivazidis S, High BA, Buscaglia G, Corrigan
T, Valdez R, et al: Neuron-specific mitochondrial oxidative stress
results in epilepsy, glucose dysregulation and a striking astrocyte
response. Neurobiol Dis. 158:1054702021. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Zhang S, Chen F, Zhai F and Liang S: Role
of HMGB1/TLR4 and IL-1β/IL-1R1 signaling pathways in epilepsy.
Front Neurol. 13:9042252022. View Article : Google Scholar
|
|
146
|
Kim JE and Kang TC: Differential roles of
mitochondrial translocation of active caspase-3 and HMGB1 in
neuronal death induced by status epilepticus. Front Cell Neurosci.
12:3012018. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Hyun HW, Ko AR and Kang TC: Mitochondrial
translocation of high mobility group box 1 facilitates LIM kinase
2-mediated programmed necrotic neuronal death. Front Cell Neurosci.
10:992016. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Kim JE, Park H, Kim TH and Kang TC: LONP1
regulates mitochondrial accumulations of HMGB1 and Caspase-3 in CA1
and PV neurons following status epilepticus. Int J Mol Sci.
22:22752021. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Pauletti A, Terrone G, Shekh-Ahmad T,
Salamone A, Ravizza T, Rizzi M, Pastore A, Pascente R, Liang LP,
Villa BR, et al: Targeting oxidative stress improves disease
outcomes in a rat model of acquired epilepsy. Brain. 142:e392019.
View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Kumar P, Osahon OW and Sekhar RV: GlyNAC
(Glycine and N-Acetylcysteine) supplementation in mice increases
length of life by correcting glutathione deficiency, oxidative
stress, mitochondrial dysfunction, abnormalities in mitophagy and
nutrient sensing, and genomic damage. Nutrients. 14:11142022.
View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Mohseni-Moghaddam P, Roghani M,
Khaleghzadeh-Ahangar H, Sadr SS and Sala C: A literature overview
on epilepsy and inflammasome activation. Brain Res Bull.
172:229–235. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Abais JM, Zhang C, Xia M, Liu Q, Gehr TW,
Boini KM and Li PL: NADPH oxidase-mediated triggering of
inflammasome activation in mouse podocytes and glomeruli during
hyperhomocysteinemia. Antioxid Redox Signal. 18:1537–1548. 2013.
View Article : Google Scholar :
|
|
153
|
Subramanian N, Natarajan K, Clatworthy MR,
Wang Z and Germain RN: The adaptor MAVS promotes NLRP3
mitochondrial localization and inflammasome activation. Cell.
153:348–361. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Zhou R, Yazdi AS, Menu P and Tschopp J: A
role for mitochondria in NLRP3 inflammasome activation. Nature.
469:221–225. 2011. View Article : Google Scholar
|
|
155
|
Rong S, Wan D, Fan Y, Liu S, Sun K, Huo J,
Zhang P, Li X, Xie X, Wang F and Sun T: Amentoflavone affects
epileptogenesis and exerts neuroprotective effects by inhibiting
NLRP3 inflammasome. Front Pharmacol. 10:8562019. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Liu Z, Xian H, Ye X, Chen J, Ma Y and
Huang W: Increased levels of NLRP3 in children with febrile
seizures. Brain Dev. 42:336–341. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
de Brito Toscano EC, Vieira EL, Dias BB,
Caliari MV, Gonçalves AP, Giannetti AV, Siqueira JM, Suemoto CK,
Leite RE, Nitrini R, et al: NLRP3 and NLRP1 inflammasomes are
up-regulated in patients with mesial temporal lobe epilepsy and may
contribute to overexpression of caspase-1 and IL-β in sclerotic
hippocampi. Brain Res. 1752:1472302021. View Article : Google Scholar
|
|
158
|
Tan CC, Zhang JG, Tan MS, Chen H, Meng DW,
Jiang T, Meng XF, Li Y, Sun Z, Li MM, et al: NLRP1 inflammasome is
activated in patients with medial temporal lobe epilepsy and
contributes to neuronal pyroptosis in amygdala kindling-induced rat
model. J Neuroinflammation. 12:182015. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Samidurai M, Tarale P, Janarthanam C,
Estrada CG, Gordon R, Zenitsky G, Jin H, Anantharam V, Kanthasamy
AG and Kanthasamy A: tumor necrosis factor-like weak inducer of
apoptosis (TWEAK) enhances activation of STAT3/NLRC4 inflammasome
signaling axis through PKCdelta in AStrocytes: Implications for
Parkinson's disease. Cells. 9:18312020. View Article : Google Scholar
|
|
160
|
Zadori D, Klivenyi P, Szalardy L, Fulop F,
Toldi J and Vecsei L: Mitochondrial disturbances, excitotoxicity,
neuroinflammation and kynurenines: Novel therapeutic strategies for
neurodegenerative disorders. J Neurol Sci. 322:187–191. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Prasuhn J, Davis RL and Kumar KR:
Targeting mitochondrial impairment in Parkinson's disease:
Challenges and opportunities. Front Cell Dev Biol. 8:6154612020.
View Article : Google Scholar
|
|
162
|
Vos M, Esposito G, Edirisinghe JN, Vilain
S, Haddad DM, Slabbaert JR, Van Meensel S, Schaap O, De Strooper B,
Meganathan R, et al: Vitamin K2 is a mitochondrial electron carrier
that rescues pink1 deficiency. Science. 336:1306–1310. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Bronzuoli MR, Iacomino A, Steardo L and
Scuderi C: Targeting neuroinflammation in Alzheimer's disease. J
Inflamm Res. 9:199–208. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Fu AK, Hung KW, Yuen MY, Zhou X, Mak DS,
Chan IC, Cheung TH, Zhang B, Fu WY, Liew FY and Ip NY: IL-33
ameliorates Alzheimer's disease-like pathology and cognitive
decline. Proc Natl Acad Sci USA. 113:E2705–E2713. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Wang X, Sun G, Feng T, Zhang J, Huang X,
Wang T, Xie Z, Chu X, Yang J, Wang H, et al: Sodium oligomannate
therapeutically remodels gut microbiota and suppresses gut
bacterial amino acids-shaped neuroinflammation to inhibit
Alzheimer's disease progression. Cell Res. 29:787–803. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Regen F, Hellmann-Regen J, Costantini E
and Reale M: Neuroinflammation and Alzheimer's disease:
Implications for microglial activation. Curr Alzheimer Res.
14:1140–1148. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
La Torre ME, Cianciulli A, Monda V, Monda
M, Filannino FM, Antonucci L, Valenzano A, Cibelli G, Porro C,
Messina G, et al: α-Tocopherol protects
lipopolysaccharide-activated BV2 microglia. Molecules. 28:33402023.
View Article : Google Scholar
|
|
168
|
Shabab T, Khanabdali R, Moghadamtousi SZ,
Kadir HA and Mohan G: Neuroinflammation pathways: A general review.
Int J Neurosci. 127:624–633. 2017. View Article : Google Scholar
|
|
169
|
Heger LM, Wise RM, Hees JT, Harbauer AB
and Burbulla LF: Mitochondrial phenotypes in Parkinson's diseases-a
focus on human iPSC-derived dopaminergic neurons. Cells.
10:34362021. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Yu G, Wang Y and Zhao J: Inhibitory effect
of mitoquinone against the α-synuclein fibrillation and relevant
neurotoxicity: Possible role in inhibition of Parkinson's disease.
Biol Chem. 403:253–263. 2022. View Article : Google Scholar
|
|
171
|
Snow BJ, Rolfe FL, Lockhart MM, Frampton
CM, O'Sullivan JD, Fung V, Smith RA, Murphy MP and Taylor KM;
Protect Study Group: A double-blind, placebo-controlled study to
assess the mitochondria-targeted antioxidant MitoQ as a
disease-modifying therapy in Parkinson's disease. Mov Disord.
25:1670–1674. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Lehmann S, Loh SH and Martins LM:
Enhancing NAD(+) salvage metabolism is neuroprotective in a PINK1
model of Parkinson's disease. Biol Open. 6:141–147. 2017.
|
|
173
|
Monti DA, Zabrecky G, Kremens D, Liang TW,
Wintering NA, Bazzan AJ, Zhong L, Bowens BK, Chervoneva I, Intenzo
C and Newberg AB: N-Acetyl cysteine is associated with dopaminergic
improvement in Parkinson's disease. Clin Pharmacol Ther.
106:884–890. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Sathe AG, Tuite P, Chen C, Ma Y, Chen W,
Cloyd J, Low WC, Steer CJ, Lee BY, Zhu XH and Coles LD:
Pharmacokinetics, safety, and tolerability of orally administered
ursodeoxycholic acid in patients with Parkinson's disease-a pilot
study. J Clin Pharmacol. 60:744–750. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Bell SM, Barnes K, Clemmens H, Al-Rafiah
AR, Al-Ofi EA, Leech V, Bandmann O, Shaw PJ, Blackburn DJ,
Ferraiuolo L and Mortiboys H: Ursodeoxycholic acid improves
mitochondrial function and redistributes Drp1 in fibroblasts from
patients with either sporadic or familial Alzheimer's Disease. J
Mol Biol. 430:3942–3953. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Lin MW, Lin CC, Chen YH, Yang HB and Hung
SY: Celastrol inhibits dopaminergic neuronal death of Parkinson's
disease through activating mitophagy. Antioxidants (Basel).
9:372019. View Article : Google Scholar
|
|
177
|
Bido S, Soria FN, Fan RZ, Bezard E and
Tieu K: Mitochondrial division inhibitor-1 is neuroprotective in
the A53T-α-synuclein rat model of Parkinson's disease. Sci Rep.
7:74952017. View Article : Google Scholar
|
|
178
|
Curtis WM, Seeds WA, Mattson MP and
Bradshaw PC: NADPH and mitochondrial quality control as targets for
a circadian-based fasting and exercise therapy for the treatment of
Parkinson's disease. Cells. 11:24162022. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Ferreira AFF, Binda KH, Singulani MP,
Pereira CPM, Ferrari GD, Alberici LC, Real CC and Britto LR:
Physical exercise protects against mitochondria alterations in the
6-hidroxydopamine rat model of Parkinson's disease. Behav Brain
Res. 387:1126072020. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Kappos L, Bar-Or A, Cree BAC, Fox RJ,
Giovannoni G, Gold R, Vermersch P, Arnold DL, Arnould S, Scherz T,
et al: Siponimod versus placebo in secondary progressive multiple
sclerosis (EXPAND): A double-blind, randomised, phase 3 study.
Lancet. 391:1263–1273. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Montalban X, Hauser SL, Kappos L, Arnold
DL, Bar-Or A, Comi G, de Seze J, Giovannoni G, Hartung HP, Hemmer
B, et al: Ocrelizumab versus placebo in primary progressive
multiple sclerosis. N Engl J Med. 376:209–220. 2017. View Article : Google Scholar
|
|
182
|
Lublin F, Miller DH, Freedman MS, Cree
BAC, Wolinsky JS, Weiner H, Lubetzki C, Hartung HP, Montalban X,
Uitdehaag BMJ, et al: Oral fingolimod in primary progressive
multiple sclerosis (INFORMS): A phase 3, randomised, double-blind,
placebo-controlled trial. Lancet. 387:1075–1084. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Waxman SG: Axonal conduction and injury in
multiple sclerosis: The role of sodium channels. Nat Rev Neurosci.
7:932–941. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Picone P and Nuzzo D: Promising treatment
for multiple sclerosis: Mitochondrial transplantation. Int J Mol
Sci. 23:22452022. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Moos WH, Faller DV, Glavas IP, Kanara I,
Kodukula K, Pernokas J, Pernokas M, Pinkert CA, Powers WR, Sampani
K, et al: Epilepsy: Mitochondrial connections to the 'Sacred'
disease. Mitochondrion. 72:84–101. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Silvestro S, Mammana S, Cavalli E,
Bramanti P and Mazzon E: Use of cannabidiol in the treatment of
epilepsy: Efficacy and security in clinical trials. Molecules.
24:14592019. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Ramirez A, Old W, Selwood DL and Liu X:
Cannabidiol activates PINK1-Parkin-dependent mitophagy and
mitochondrial-derived vesicles. Eur J Cell Biol. 101:1511852022.
View Article : Google Scholar :
|
|
188
|
Bhunia S, Kolishetti N, Arias AY, Vashist
A and Nair M: Cannabidiol for neurodegenerative disorders: A
comprehensive review. Front Pharmacol. 13:9897172022. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Britch SC, Babalonis S and Walsh SL:
Cannabidiol: Pharmacology and therapeutic targets.
Psychopharmacology (Berl). 238:9–28. 2021. View Article : Google Scholar
|
|
190
|
Aledo-Serrano A, Hariramani R,
Gonzalez-Martinez A, Álvarez-Troncoso J, Toledano R, Bayat A,
Garcia-Morales I, Becerra JL, Villegas-Martínez I,
Beltran-Corbellini A and Gil-Nagel A: Anakinra and tocilizumab in
the chronic phase of febrile infection-related epilepsy syndrome
(FIRES): Effectiveness and safety from a case-series. Seizure.
100:51–55. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Stienen MN, Haghikia A, Dambach H, Thöne
J, Wiemann M, Gold R, Chan A, Dermietzel R, Faustmann PM, Hinkerohe
D and Prochnow N: Anti-inflammatory effects of the anticonvulsant
drug levetiracetam on electrophysiological properties of astroglia
are mediated via TGFβ1 regulation. Br J Pharmacol. 162:491–507.
2011. View Article : Google Scholar :
|
|
192
|
Stockburger C, Miano D, Baeumlisberger M,
Pallas T, Arrey TN, Karas M, Friedland K and Müller WE: A
mitochondrial role of sv2a protein in aging and Alzheimer's
disease: Studies with levetiracetam. J Alzheimers Dis. 50:201–215.
2016. View Article : Google Scholar
|
|
193
|
Yang N, Guan QW, Chen FH, Xia QX, Yin XX,
Zhou HH and Mao XY: Antioxidants targeting mitochondrial oxidative
stress: Promising neuroprotectants for epilepsy. Oxid Med Cell
Longev. 2020:66871852020. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Fields M, Marcuzzi A, Gonelli A, Celeghini
C, Maximova N and Rimondi E: Mitochondria-Targeted antioxidants, an
innovative class of antioxidant compounds for neurodegenerative
diseases: Perspectives and limitations. Int J Mol Sci. 24:37392023.
View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Huenchuguala S and Segura-Aguilar J:
Single-neuron neurodegeneration as a degenerative model for
Parkinson's disease. Neural Regen Res. 19:529–535. 2024. View Article : Google Scholar
|
|
196
|
Leitao-Rocha A, Guedes-Dias P, Pinho BR
and Oliveira JM: Trends in mitochondrial therapeutics for
neurological disease. Curr Med Chem. 22:2458–2467. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Yoshinaga N and Numata K: Rational designs
at the forefront of mitochondria-targeted gene delivery: Recent
progress and future perspectives. ACS Biomater Sci Eng. 8:348–359.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Silva-Pinheiro P and Minczuk M: The
potential of mitochondrial genome engineering. Nat Rev Genet.
23:199–214. 2022. View Article : Google Scholar
|