Introduction
Clear cell renal cell carcinoma (ccRCC), the most
common renal malignancy, originates from renal tubular epithelial
cells. ccRCC is the most common histological subtype of RCC,
accounting for 80–90% of all RCC cases (1). In addition, patients with ccRCC have a
poor prognosis. Surgical treatment is effective for patients with
ccRCC at an early stage; however, recurrence and metastasis may
occur in ≤30% of patients following radical surgery, resulting in
poor prognosis (2).
Enhancers are DNA fragments that normally range from
a few hundred to a few thousand base pairs in length and are
typically occupied by several transcription factors (3,4). In
addition to enhancers, large stretches of tightly linked enhancers,
known as super-enhancers (SEs), exist in the genome. SEs serve as a
‘platform’ for controlling the spatiotemporal expression of genes
by converging internal and external environmental signaling
pathways (5). Studies have
identified close relationships between SEs and tumor processes such
as epithelial-mesenchymal transition (6), metabolic reprogramming (7), immune escape (8) and resistance to apoptosis (9).
The treatment for RCC has rapidly advanced in recent
years. Studies have indicated that RCC may be immunogenic (10) and sensitive to immunotherapy
(11). Immunotherapy and targeted
therapy have broadened the options for treating ccRCC; however,
some patients with ccRCC first show symptoms after the cancer cells
have metastasized, and these patients often have a 5-year survival
rate of <20% (12). Studies have
found that tumor cells upregulate the expression of programmed
death-ligand 1 (PD-L1) during transcription and translation, and
that there is an important SE, PD-L1/L2-SE, between the coding
regions of PD-L1 and PD-L2 (13,14).
PD-L1/L2-SE promotes the expression of PD-L1 and PD-L2, achieving
immune escape by inhibiting CD8+ T cell activation; by
contrast, its absence can cause tumor cells to lose their immune
escape ability and become sensitive to T cell killing (15). This mechanism has been found in
numerous tumor types, such as thyroid papillary carcinoma and
gastric, lung and breast cancer (16). In addition, a study has indicated
that treatment of colorectal cancer cell lines with the SE
inhibitors, JQ-1 and ibet-151, could lead to inhibition of cell
proliferation and downregulation of interleukin-20 receptor α
(IL-20RA) expression (17).
Following knockdown of the IL-20RA gene driven by SEs, the ability
of colorectal cancer cells to proliferate, migrate and invade in
vitro was markedly reduced. These findings suggest that
SE-related IL-20RA may inhibit the immune response in colorectal
cancer, and that a close relationship exists between SEs and immune
escape. Therefore, exploring the relationship between SEs and
immune genes may provide novel ideas for ccRCC immunotherapy.
In the present study, an Arraystar human SE-long
non-coding RNA (lncRNA) microarray was first performed using paired
ccRCC and peritumoral tissues to identify SE-related genes. The
overlap of these genes with immune genes was determined to identify
SE-related immune genes. A model for predicting clinical prognosis
and response to immunotherapy was built following the comprehensive
analysis of a ccRCC gene expression dataset from The Cancer Genome
Atlas (TCGA) database to identify SE-related immune genes. In
addition, based on the constructed risk model, tumor immune
response, mutation load and drug sensitivity in ccRCC were
explored. These findings may provide a potential strategy for the
prognostic evaluation and treatment of patients with ccRCC.
Materials and methods
Patients and tissue samples
The present study was approved by The Ethics
Committee of the First Affiliated Hospital of Harbin Medical
University (Harbin, China; approval no. 201734). The research was
conducted in accordance with the Declaration of Helsinki. This
study included a total of 5 patients with ccRCC (2 female patients
and 3 male patients, aged between 45 and 60 years old). All 5
patients were first diagnosed by postoperative pathology. Written
informed consent was obtained from all patients. A total of 5 pairs
of ccRCC tissues and adjacent cancerous tissues were obtained. All
of the tumor and adjacent normal tissues were collected from
surgically removed specimens at The First Affiliated Hospital of
Harbin Medical University in April and May 2017. The samples were
snap frozen in liquid nitrogen and then stored at −80°C until
analysis.
Microarray analysis and the collection
of public data
The microarray and bioinformatic analyses of the 5
patients with ccRCC were conducted by Arraystar, Inc. Sample
labeling and array hybridization were performed according to the
Agilent One-Color Microarray-Based Gene Expression Analysis
protocol (Agilent Technologies, Inc) with minor modifications.
Briefly, mRNA was purified from total RNA following the removal of
rRNA using an mRNA-ONLY™ Eukaryotic mRNA Isolation Kit (RNeasy Mini
Kit; cat. no. 74104; Qiagen, Inc.). Next, each sample was amplified
and transcribed into fluorescent cRNA along the entire length of
the transcripts without a 3′ bias using a random priming method at
4°C and the Arraystar Flash RNA Labeling Kit (Arraystar Inc.). The
labeled cRNAs were purified using an RNeasy Mini Kit (Qiagen,
Inc.). The concentration and specific activity of the labeled cRNAs
(pmol Cy3/µg cRNA) were measured using a NanoDrop ND-1000. A total
of 50 µl hybridization solution was dispensed into the gasket slide
and assembled to the lncRNA expression microarray slide (Agilent
Gene Expression Hybridization Kit (cat. no. 5188-5242; Agilent
Technologies, Inc.). The slides were incubated for 17 h at 65°C in
an Agilent Hybridization Oven. The hybridized arrays were washed
with Milli-Q water and scanned using the Agilent G2505C DNA
Microarray Scanner. Agilent Feature Extraction software (version
11.0.1.1; Agilent Technologies, Inc.) was used to analyze the
acquired array images. Quantile normalization and subsequent data
processing were performed using the GeneSpring GX v12.1 software
package (Agilent Technologies, Inc.). Results with |Log2 fold
change|≥2.0 and P≤0.05 were considered as differentially expressed
SE-lncRNAs and mRNAs. The resulting SE-lncRNA microarray data were
deposited in the Gene Expression Omnibus database (accession no.
GSE249053).
Data on immune genes including gene names,
chromosomal location and classification, were retrieved from the
IMMPORT database (https://www.immport.org/home). The transcriptome
RNA-sequencing, clinical and mutation data of patients with ccRCC
were downloaded from TCGA (https://portal.gdc.cancer.gov), and the public data
were obtained on November 23, 2022. R software (version 4.2.2;
http://www.r-project.org/) and
Bioconductor packages (http://www.bioconductor.org/) were used for data
analysis. The workflow of the present study is shown in Fig. 1.
Identification of immune genes
associated with SEs
A total of 1,501 differentially expressed SE-related
RNAs were screened using microarray analysis. In addition, 2,483
immune-related genes were obtained from the IMMPORT database. By
intersecting these two datasets, 112 overlapping genes were
identified as SE-related immune genes. Subsequently, univariate Cox
regression analysis of overall survival (OS) was performed to
screen for SE-related immune genes with prognostic value using TCGA
data. Finally, 43 SE-related immune genes were identified which
were significantly associated with the prognosis of patients with
ccRCC.
Construction of a risk model using
SE-related immune genes
The sorted data from TCGA were randomly divided into
training and testing sets. Multivariate Cox regression analysis was
performed using the training set, which led to the identification
of 6 SE-related immune genes. The risk model was constructed
according to the degree of expression and coefficient of these 6
genes, and was termed ‘SIRM’. The risk score was calculated using
the following formula: Risk score = expr (RNA1) × coef (RNA1) +
expr (RNA2) × coef (RNA2) +……+ expr (RNAn) × coef (RNAn), where
expr indicates gene expression and coef indicated coefficient. The
model was applied to every sample in the training and testing
datasets, and the median risk score value was set as the cut off;
thus, samples in both datasets were divided into low- and high-risk
subgroups.
Principal component analysis
(PCA)
PCA can effectively reduce the dimensions of
high-dimensional data and visualize grouping (18). PCA was conducted based on the 6
genes used to build the risk model. PCA was performed on whole gene
expression profiles, SE-related immune genes and risk models.
Kaplan-Meier survival analysis and
independence of the SIRM model
The prognosis of patients was scored using the risk
model. Based on the median risk score, patients with ccRCC were
divided into high- and low-risk groups. Kaplan-Meier survival
analysis was performed to evaluate differences in OS between the
high- and low-risk groups using the R packages, ‘survMiner’
(version 0.4.9) and ‘Survival’ (version 3.4.0). Both groups were
analyzed using univariate and multivariate Cox regression to
determine whether the prognostic pattern was an independent
variable based on age, sex, stage and grade (19).
Preliminary research on tumor mutation
burden (TMB) and immunotherapy
The mutation data obtained from TCGA were processed
and calculated using the R package, ‘maftools’ (version 2.14.0). To
evaluate the gene mutation level in ccRCC, TMB was determined
according to somatic mutation data from TCGA database. The Tumor
Immune Dysfunction and Exclusion (TIDE) algorithm was employed to
predict the possibility of an immunotherapy response (20).
Predicting OS by building a
nomogram
To predict 1-, 3- and 5-year OS, a nomogram with
predictive ability was established using the R packages, ‘Regplot’
(version 1.1), ‘Survival’ (version 3.4.0) and ‘Rms’ (version
6.3.0). Subsequently, to show consistency between the results of
practical application and risk model prediction, correction curves
based on the Hosmer-Lemeshow test were chosen.
Gene ontology (GO) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of
SE-related immune genes
To explore the biological properties of SE-related
immune genes, GO and KEGG enrichment analysis were conducted using
the R package, ‘clusterprofiler’ (version 4.6.0).
Investigation of SIRM-targeting
molecules for clinical application
To explore potential therapeutic drugs for the
treatment of patients with ccRCC, the half-maximal inhibitory
concentration (IC50) of drugs from the Genomics of Drug
Sensitivity in Cancer (GDSC) website (https://www.cancerrxgene.org/) and ccRCC data were
calculated using the R package, ‘pRRophetic’.
Statistical analysis
Data processing and bioinformatics analysis were
conducted using R (version 4.2.2; http://www.r-project.org/) and Perl Data Language
(https://www.perl.org/). The Wilcoxon signed-rank
test was performed to compare the indicators of ccRCC in tissue and
control samples. |Pearson R|>0.4. P<0.05 was considered to
indicate a statistically significant difference.
Results
A constructed risk model based on
SE-related immune genes can predict the prognosis of patients with
ccRCC
In the present study, a total of 112 differentially
expressed genes were identified as SE-related immune genes and
subsequently included in a univariate Cox regression analysis. The
results indicated that 43 differentially expressed SE-related
immune genes were significantly associated with OS (Fig. 2A). Then Lasso-penalized Cox
regression was performed using the 43 genes to construct the risk
model. Finally, 6 SE-related immune genes were found to be
significantly associated with OS (Table
I) and were used to construct the risk model (Fig. 2B and C), with risk
score=0.394961526106691 × expr[erythropoietin receptor (EPOR)]
+0.766612172033089 × expr[BH3 interacting domain death agonist
(BID)] +0.508670285309477 × expr[γ-interferon-inducible lysosomal
thiol reductase (IFI30)] +0.180571536803486 × expr(ISG15)
−0.283424379032887 × expr[platelet derived growth factor D (PDGFD)]
−0.54604544798391 × expr[XC motif chemokine receptor 1 (XCR1)].
 | Table I.Genes names used to build the risk
model and their coefficients. |
Table I.
Genes names used to build the risk
model and their coefficients.
| Gene name | Coefficient |
|---|
| EPOR |
0.394961526106691 |
| BID |
0.766612172033089 |
| IFI30 |
0.508670285309477 |
| ISG15 |
0.180571536803486 |
| PDGFD |
−0.283424379032887 |
| XCR1 |
−0.54604544798391 |
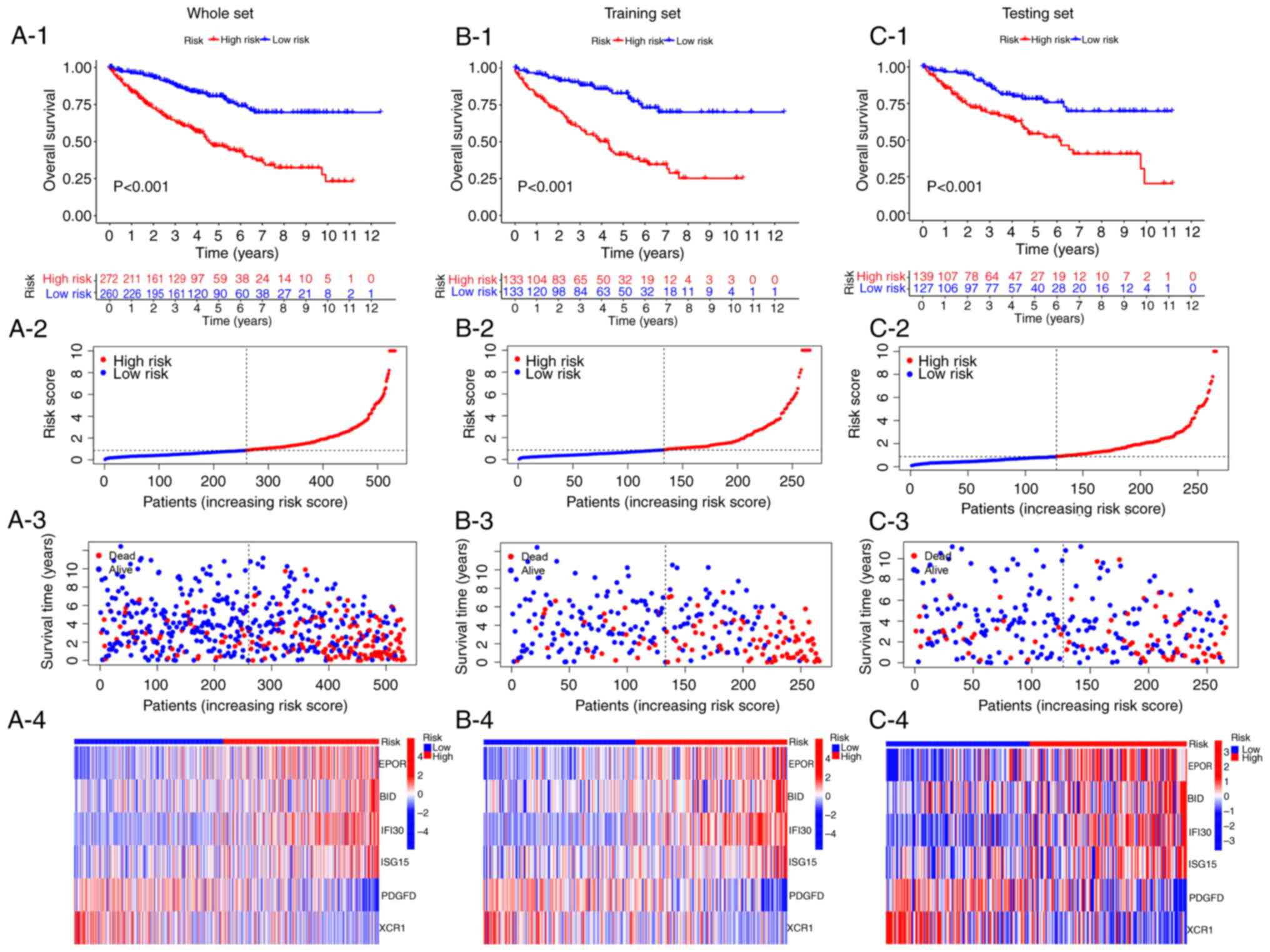
Kaplan-Meier survival analysis was conducted using
the whole, training and testing sets. The results demonstrated that
the low-risk subgroup had a longer survival time than the high-risk
subgroup, and the mortality was higher in the high-risk subgroup
compared with the low-risk subgroup (Fig. 3). Regarding the OS of the whole set,
the low-risk subgroup had a significantly longer OS than the
high-risk subgroup (P<0.001; Fig.
3A-1). As the risk score increases, the mortality rate of
patients with ccRCC also increases (P<0.001; Fig. 3A-2 and A-3). Consistently, in the
training and testing sets, the OS in the high-risk subgroup was
shorter than the low-risk subgroup (P<0.001; Fig. 3B-1 and C-1). And the mortality rate
also increases with the risk score increases in the training and
testing sets. The distribution of the vital status and survival
time of the patients with ccRCC according to the risk score in the
training set are displayed in Fig. 3B-2
and B-3; meanwhile, the vital status and survival time of the
testing set are shown in Fig. 3C-2 and
C-3. In addition, a similar pattern was observed in the
heatmaps of the 6 SE-related immune genes in the whole, training
and testing sets. Four genes with positive coefficients are highly
expressed in the high-risk group, while two genes with negative
coefficients are highly expressed in the low-risk group (Fig. 3A-4, B-4 and C-4).
High-risk group has a worse prognosis
and pathological characteristics than the low-risk group
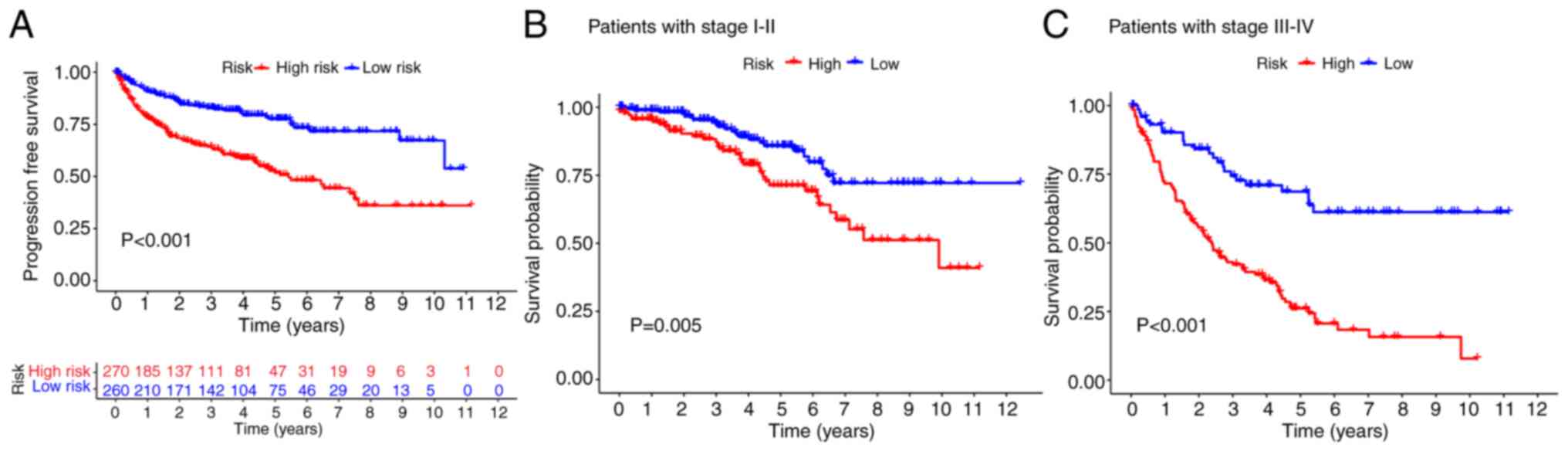
The high-risk group had a lower progression-free
survival rate than the low-risk group (Fig. 4A). The survival probability based on
tumor stage was analyzed to verify the validity of the model in the
context of other variables. The results revealed that the high-risk
group had a decreased OS rate compared with the low-risk group
(Fig. 4B and C). These outcomes
confirmed that the model could be applied to various clinical
factors.
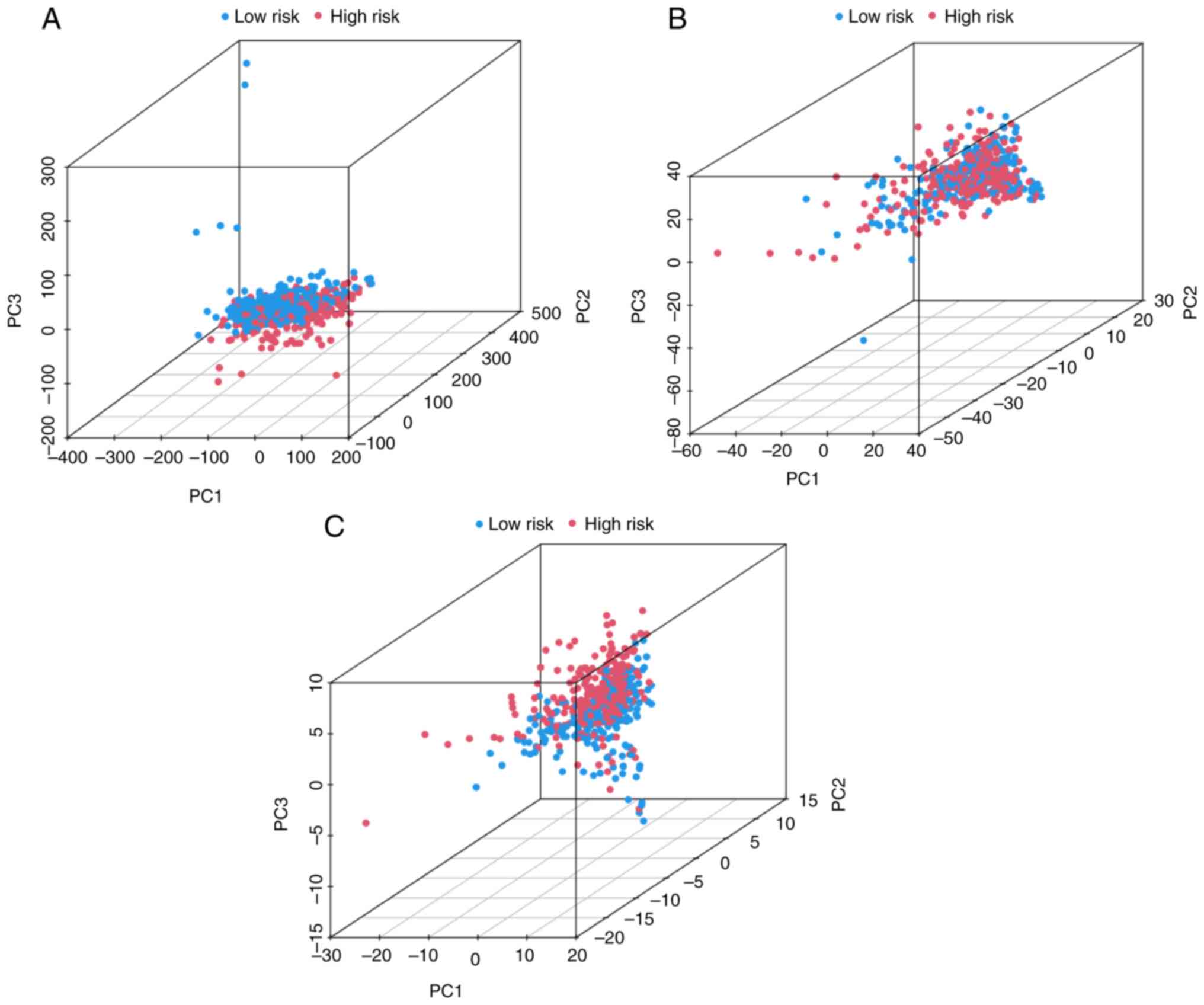
PCA indicates that the high- and
low-risk groups can be distinguished by the 6 SE-related immune
genes of SIRM
To verify the grouping ability of SIRM, PCA was
conducted according to the expression of the whole genes,
SE-related immune genes and the 6 SE-related immune genes of the
risk model (Fig. 5A-C,
respectively). The distributions of the high- and low-risk groups
were distinguishable and relatively convergent (Fig. 5A and B). However, the results
obtained using SIRM revealed that patients in the high- and
low-risk groups were significantly distinguished (Fig. 5C), indicating that the 6 SE-related
immune genes used to construct the risk model were able to
distinguished high- and low-risk patients.
High-risk group has a higher TMB and a
more sensitive immunotherapy response
Mutation information was stratified using the R
software tool, ‘maftools’. The top 15 genes with the highest
mutation frequencies in the high- and low-risk groups are shown as
waterfall plots in Fig. 6A and B.
TMB values were calculated using data from TGCA somatic mutations.
The high-risk group had a higher number of cancer mutations than
the low-risk group (Fig. 6C).
Kaplan-Meier survival analysis was performed using the TMB data
(Fig. 6D and E), and it was shown
that the low-mutation group had a significantly higher survival
probability than the high-mutation group (Fig. 6D). Patients with high mutation rates
in the high-risk group had the lowest survival probability, whereas
those with low mutation rates in the low-risk group had the highest
survival probability (Fig. 6E).
Subsequently, the relationship between SIRM and response to
immunotherapy was explored. The results indicated that the
high-risk group had a higher response to immunotherapy, indicating
that SIRM could serve as a model to predict TIDE (Fig. 6F).
GO and KEGG enrichment analysis of
SE-related immune genes
GO and KEGG enrichment analyses were performed on
the SE-related immune genes using the R package, ‘clusterprofiler’
(Fig. 7) (21). Under biological processes,
SE-related immune genes significantly contributed to the ‘defense
response to bacterium’, ‘negative regulation of hydrolase activity’
and ‘humoral immune response’. Regarding cellular components,
‘collagen-containing extracellular matrix’, ‘blood microparticle’
and ‘specific granule lumen’ were significantly abundant.
SE-related immune genes were enriched for molecular functions
associated with ‘signaling receptor activator activity’, ‘receptor
ligand activity’ and ‘enzyme inhibitor activity’ (Fig. 7B). These findings suggested that the
SE-related immune genes have major roles in the evolution of immune
responses. Moreover, KEGG analysis revealed that SE-related immune
genes were enriched in the ‘cytokine-cytokine receptor
interaction’, ‘protein digestion and absorption’ and ‘viral protein
interaction with cytokine and cytokine receptor’ (Fig. 7C and D).
SIRM can independently predict the
prognosis of patients with ccRCC
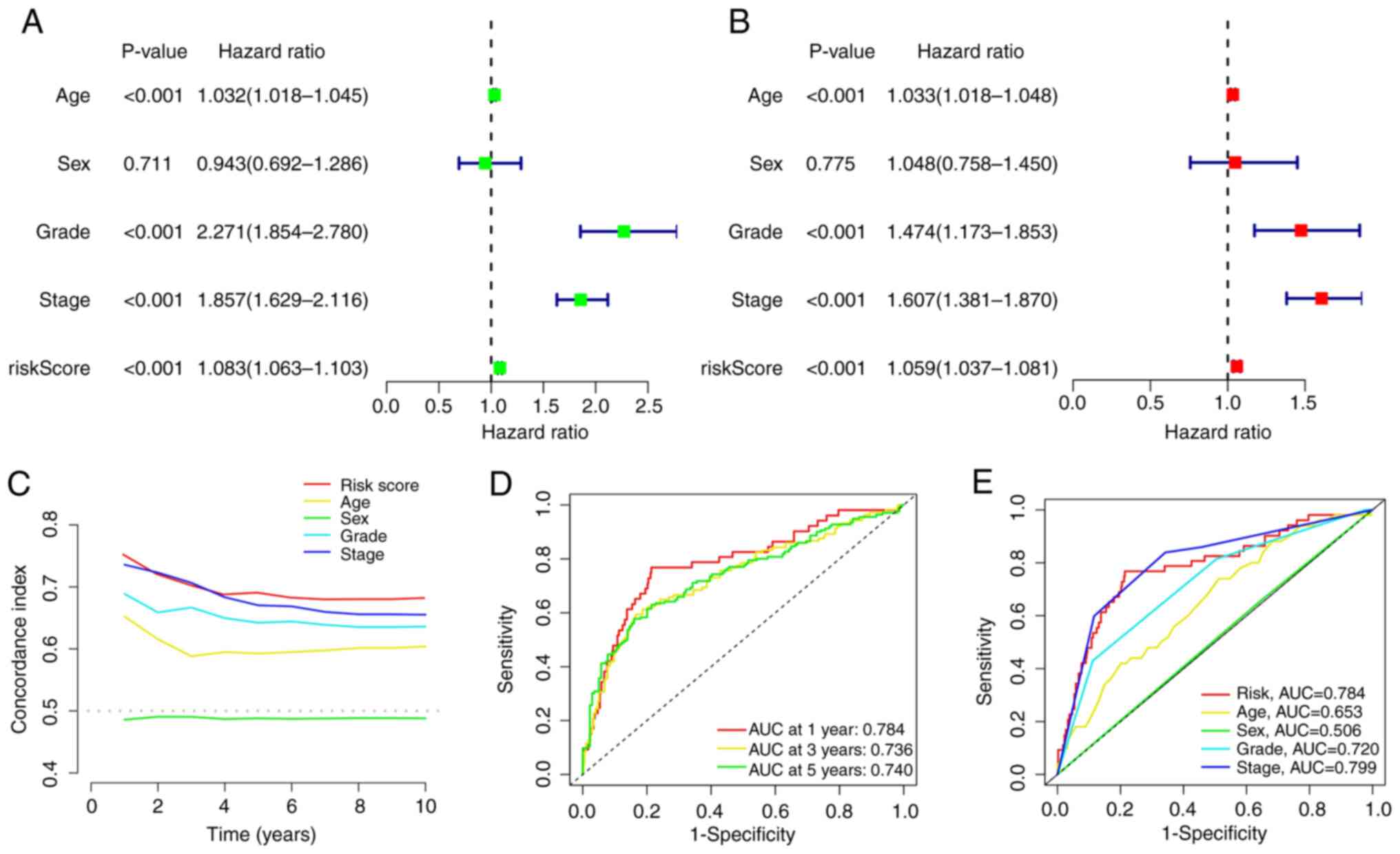
Univariate and multivariate Cox regression analyses
were performed to assess the independence of the risk model. The
results of the univariate regression analysis showed that the
hazard ratio (HR) was 1.083 and the 95% confidence interval (CI)
was 1.063–1.103 (P<0.001; Fig.
8A). The results of the multivariate regression analysis showed
that the HR was 1.059 and the 95% CI was 1.037–1.081 (P<0.001;
Fig. 8B). To further evaluate the
independence and sensitivity of the risk model in predicting
prognoses, the concordance index (C-index) and area under the
receiver operating characteristic (ROC) curve (AUC) for the risk
model were calculated. The results indicated that the C-index of
the risk score remained high, which was consistent with the general
trend (Fig. 8C). In addition, the
AUCs for 1 year, 3 years and 5 years of the risk model were
>0.7, and the AUC of the risk model was notably higher than that
of the other prognostic factors (Fig.
8D and E). This result provided further evidence that SIRM can
accurately predict prognosis even in the absence of other clinical
indicators.
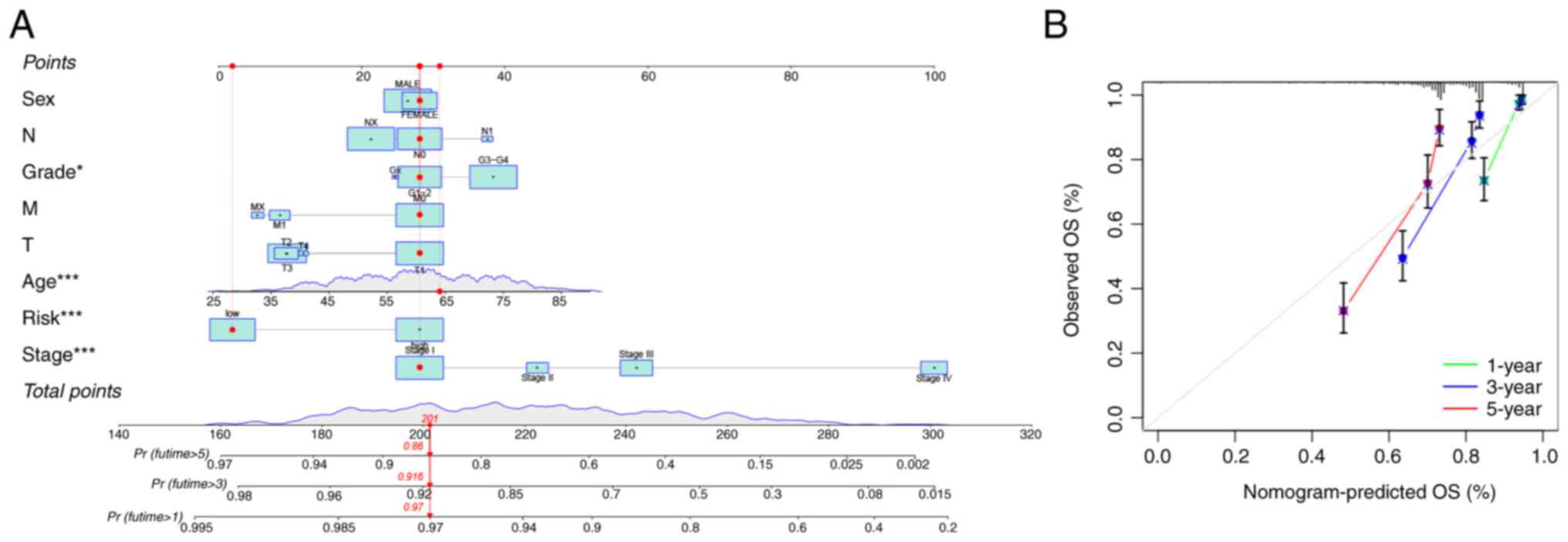
Establishment of a nomogram to predict
the survival of patients with ccRCC
To predict OS at 1, 3 and 5 years, a nomogram was
constructed that incorporated risk scores and clinical features
such as sex, age, stage and grade. The nomogram was used to predict
patient survival based on these clinical features and the risk
scores (Fig. 9A). The calibration
charts for 1 year, 3 years and 5 years showed good consistency
between the predicted survival probability of the nomogram and the
actual survival results (Fig.
9B).
Screening of potential treatment drugs
for ccRCC
Potential treatment drugs were also explored using
SIRM for treating patients with ccRCC using the R package
‘pRRophetic’ (version 0.5). Finally, 61 potential drugs were
screened, and the IC50 values of these drugs were
significantly different between the two risk groups. The four
potentially sensitive drugs displaying the lowest IC50
values are shown in Fig. 10.
Docetaxel, SN-38 and vinblastine may be the most suitable for
patients in the high-risk group, whereas pazopanib may be
beneficial for patients in the low-risk group.
Discussion
RCC is one of the most prevalent cancer types of the
urinary system, and its prevalence has been on the increase
(22). RCC is insensitive to
radiotherapy and chemotherapy, and drug resistance may occur in
patients treated with targeted therapy. This leads to a poor
prognosis for patients with RCC (23,24).
SEs have a key role in carcinogenesis, and numerous studies have
revealed that SEs play essential roles in immune evasion in breast
cancer (15), stomach
adenocarcinoma (25) and colorectal
cancer (17). In addition, studies
revealed that conventional immunotherapies, such as IFN-α and IL-2,
prolonged OS. However, the duration of their responses was
restricted, and only a few patients had complete response (26,27).
Therefore, it is important to explore the roles of SE-related
immune genes in tumorigenesis and disease progression and prognosis
in order to explore potential drugs regulating this process.
In the present study, the expression of SE-RNAs in
paired malignant and adjacent normal kidney tissues from 5 patients
with ccRCC was investigated using microarray analysis. A total of
1,501 ccRCC-associated differentially expressed SE-RNAs were
identified. These SE-RNAs were intersected with 2,483 previously
identified immune genes, and 112 SE-related immune genes were
obtained. Finally, a risk model based on 6 of these SE-related
immune genes that independently predicted prognosis was
constructed.
The 6 SE-related immune genes used to build the risk
model included EPOR, BID, IFI30, ISG15, PDGFD and XCR1. EPOR has
been shown to be abnormally expressed in various cancer types, such
as breast cancer (28), acute
lymphoblastic leukemia (29) and
RCC (30). Particularly in RCC, the
induction of erythropoietin may accelerate the proliferation of RCC
cell lines in either a hypoxia-inducible factor-1α-dependent or
-independent manner (30). The
abnormal expression of BID in various digestive tumors has been
confirmed (31,32). A recent study demonstrated that
IFI30 was highly expressed in breast cancer tissues and was
associated with a poor outcome in patients. In this study, the
knockdown of IFI30 inhibited the proliferation, migration and
invasion of breast cancer cells (33). The ISG15 protein, encoded by the
ISG15 gene, is a member of the ubiquitin-like protein family and is
involved in multiple key cellular processes, including autophagy,
exosome secretion, DNA repair, immune regulation, and cancer
occurrence and progression (34).
Through in vivo and in vitro experiments, a study
demonstrated that ISG15 induces CD4 T cell proliferation and
invalidity and immune responses against tumors (35). In the present study, Kaplan-Meier
analysis was conducted to explore the relationship between the
aforementioned genes and ccRCC. As expected, EPOR, BID, IFI30 and
ISG15 were expressed at high levels in the high-risk group. A study
demonstrated that breast cancer cells (MDA-MB-231 cells) with
PDGF-D silencing had a significantly diminished aggressive
migration and invasion potential compared with other cells (SK-BR-3
and MCF7 cell lines) (36). In
addition, in vivo experiments also demonstrated that PDGF-D
silencing inhibited tumor growth and improved the survival rate of
tumor-bearing mice. Another study found that silencing XCR1
promoted hepatocellular carcinoma cell migration and invasion in
vitro and overexpressing XCR1 had an inhibitory effect
(37). In the present study, it was
shown that these two genes, PDGFD and XCR1, were expressed at low
levels in the high-risk. Therefore, the biological behavior of the
aforementioned 6 genes in tumors is consistent with the results of
the present study on ccRCC.
In the present study, patients were divided into
high- and low-risk groups based on the median score derived from
the risk model. The Kaplan-Meier survival analysis revealed that
the low-risk group had a better survival probability than the
high-risk group. According to the multivariate Cox regression
analysis, the risk model could be used as an independent risk
factor for ccRCC. In addition, the ROC curve analysis results
indicated that the risk model had more advantages than other
clinical factors in predicting the OS of patients with ccRCC. To
predict the 1-, 3- and 5-year OS rates of patients, a nomogram that
could comparatively predict patient survival was constructed. This
was beneficial for improving the validity of the risk model.
Additionally, to provide a novel avenue for
immunotherapy, the TIDE algorithm was adopted to explore
sensitivity to immunotherapy. According to these findings, the
high-risk group demonstrated a more robust immune response than the
low-risk group. This result suggested that patients with ccRCC in
the high-risk group may have a better outcome when treated with
immunotherapy. Based on this, four potential drugs with
IC50 values that differed significantly between the
high- and low-risk groups were screened out. Related studies have
demonstrated the positive effects of docetaxel, SN-38 and pazopanib
in the treatment of ccRCC (38–40).
The effectiveness of vinblastine has also been validated in animal
and cell experiments (41). These
findings therefore provide a new avenue for chemotherapy drug
selection.
A number of factors such as tumor stage and grade,
age and metastasis, affect the prognosis of patients with cancer.
However, none of these prognostic factors can accurately predict
patient survival. Thus, it is crucial to investigate predictors
that are more comprehensive, specific and accurate. SE-related
immune gene models complement the inadequacy of clinical
indicators. They also provide a new direction for exploring the
carcinogenic mechanism of ccRCC. Perhaps the potential mechanism
between the abnormal expression of immune genes regulated by SEs
and the occurrence of ccRCC can be explored. The present study
validated and determined multiple aspects of a SE-related immune
gene risk model. Therefore, this model may be flexibly applied to
predict survival probability in patients with ccRCC.
However, the present study did also have some
limitations. First, although the data on SE-related genes were
obtained through microarray analysis, the data used for model
construction and validation were sourced from a public database.
Furthermore, most of the results originated from bioinformatics
engineering. Therefore, a number of experiments are required to
verify the results of the present study and the underlying
mechanism of abnormal expression of SE-related immune genes leading
to a poor prognosis in ccRCC still needs further experimental
exploration. Second, the selected drugs identified in the present
study are only potential therapeutic drugs, and their therapeutic
effects as well as their underlying mechanism still need to be
verified. The aim is to conducted this investigation in future
studies.
In summary, the present study provided a potential
index for predicting the survival probability of patients with
ccRCC and presented a research direction into the mechanisms by
which SE-related immune genes influence the prognosis of patients
with ccRCC. Several potential drugs were screened and potential
leads for immunotherapy of ccRCC were provided. These findings may
provide a potential strategy for the prognostic evaluation and
treatment of patients with ccRCC in the future.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be found
in the Gene Expression Omnibus database under accession number
GSE249053 or at the following URL: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE249053.
Authors' contributions
ZB contributed to experimental studies, manuscript
editing and statistical analysis. JZ contributed to acquisition of
data and manuscript editing. YM contributed to data analysis. QG
and HD contributed to the literature search and analysis and
interpretation of data. BJ and FY contributed to data acquisition.
HZ and ZZ contributed to statistical analysis. ES and XG
contributed to manuscript review and made substantial contributions
to conception and design. All authors read and approved the final
version of the manuscript. ZB and ES confirm the authenticity of
all the raw data.
Ethics approval and consent to
participate
The study was approved by The Ethics Committee of
First Affiliated Hospital of Harbin Medical University (Harbin,
China; approval no. 201734). The research was conducted in
accordance with the Declaration of Helsinki. Written informed
consent was obtained from all patients for participation in the
study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ljungberg B, Albiges L, Abu-Ghanem Y,
Bensalah K, Dabestani S, Fernández-Pello S, Giles RH, Hofmann F,
Hora M, Kuczyk MA, et al: European association of urology
guidelines on renal cell carcinoma: The 2019 update. Eur Urol.
75:799–810. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Finelli A, Ismaila N, Bro B, Durack J,
Eggener S, Evans A, Gill I, Graham D, Huang W, Jewett MA, et al:
Management of small renal masses: American society of clinical
oncology clinical practice guideline. J Clin Oncol. 35:668–680.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Panne D: The enhanceosome. Curr Opin
Struct Biol. 18:236–242. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Spitz F and Furlong EE: Transcription
factors: From enhancer binding to developmental control. Nat Rev
Genet. 13:613–626. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Calo E and Wysocka J: Modification of
enhancer chromatin: What, how, and why. Mol Cell. 49:825–837. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang C, Wei S, Sun WP, Teng K, Dai MM,
Wang FW, Chen JW, Ling H, Ma XD, Feng ZH, et al:
Super-enhancer-driven AJUBA is activated by TCF4 and involved in
epithelial-mesenchymal transition in the progression of
hepatocellular carcinoma. Theranostics. 10:9066–9082. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nguyen TTT, Zhang Y, Shang E, Shu C,
Torrini C, Zhao J, Bianchetti E, Mela A, Humala N, Mahajan A, et
al: HDAC inhibitors elicit metabolic reprogramming by targeting
super-enhancers in glioblastoma models. J Clin Invest.
130:3699–3716. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Betancur PA, Abraham BJ, Yiu YY,
Willingham SB, Khameneh F, Zarnegar M, Kuo AH, McKenna K, Kojima Y,
Leeper NJ, et al: A CD47-associated super-enhancer links
pro-inflammatory signalling to CD47 upregulation in breast cancer.
Nat Commun. 8:148022017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shang E, Nguyen TTT, Shu C, Westhoff MA,
Karpel-Massler G and Siegelin MD: Epigenetic targeting of Mcl-1 is
synthetically lethal with Bcl-xL/Bcl-2 inhibition in model systems
of glioblastoma. Cancers (Basel). 12:21372020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Şenbabaoğlu Y, Gejman RS, Winer AG, Liu M,
Van Allen EM, de Velasco G, Miao D, Ostrovnaya I, Drill E, Luna A,
et al: Tumor immune microenvironment characterization in clear cell
renal cell carcinoma identifies prognostic and
immunotherapeutically relevant messenger RNA signatures. Genome
Biol. 17:2312016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Escudier B: Emerging immunotherapies for
renal cell carcinoma. Ann Oncol. 23 (Suppl 8):viii35–viii40. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dunnick NR: Renal cell carcinoma: Staging
and surveillance. Abdom Radiol (NY). 41:1079–1085. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sun C, Mezzadra R and Schumacher TN:
Regulation and function of the PD-L1 checkpoint. Immunity.
48:434–452. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu Y, Wu Y, Zhang S, Ma P, Jin X, Wang Z,
Yao M, Zhang E, Tao B, Qin Y, et al: A tumor-specific
super-enhancer drives immune evasion by guiding synchronous
expression of PD-L1 and PD-L2. Cell Rep. 29:3435–3447.e4. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ma P, Jin X, Fan Z, Wang Z, Yue S, Wu C,
Chen S, Wu Y, Chen M, Gu D, et al: Super-enhancer receives signals
from the extracellular matrix to induce PD-L1-mediated immune
evasion via integrin/BRAF/TAK1/ERK/ETV4 signaling. Cancer
Biol Med. 19:669–684. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Oh S, Shin S and Janknecht R: ETV1, 4 and
5: An oncogenic subfamily of ETS transcription factors. Biochim
Biophys Acta. 1826:1–12. 2012.PubMed/NCBI
|
|
17
|
Yu D, Yang X, Lin J, Cao Z, Lu C, Yang Z,
Zheng M, Pan R and Cai W: Super-enhancer induced IL-20RA promotes
proliferation/metastasis and immune evasion in colorectal cancer.
Front Oncol. 11:7246552021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li X, Li Y, Yu X and Jin F: Identification
and validation of stemness-related lncRNA prognostic signature for
breast cancer. J Transl Med. 18:3312020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu F, Huang X, Li Y, Chen Y and Lin L:
m6A-related lncRNAs are potential biomarkers for
predicting prognoses and immune responses in patients with LUAD.
Mol Ther Nucleic Acids. 24:780–791. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu F, Zhan X, Zheng X, Xu H, Li Y, Huang
X, Lin L and Chen Y: A signature of immune-related gene pairs
predicts oncologic outcomes and response to immunotherapy in lung
adenocarcinoma. Genomics. 112:4675–4683. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cao Y and Tang W and Tang W: Immune cell
infiltration characteristics and related core genes in lupus
nephritis: Results from bioinformatic analysis. BMC Immunol.
20:372019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pang C, Guan Y, Li H, Chen W and Zhu G:
Urologic cancer in China. Jpn J Clin Oncol. 46:497–501. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bianchi M, Gandaglia G, Trinh QD, Hansen
J, Becker A, Abdollah F, Tian Z, Lughezzani G, Roghmann F, Briganti
A, et al: A population-based competing-risks analysis of survival
after nephrectomy for renal cell carcinoma. Urol Oncol. 32:46.e1–7.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Funakoshi T, Lee CH and Hsieh JJ: A
systematic review of predictive and prognostic biomarkers for
VEGF-targeted therapy in renal cell carcinoma. Cancer Treat Rev.
40:533–547. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Peng L, Peng JY, Cai DK, Qiu YT, Lan QS,
Luo J, Yang B, Xie HT, Du ZP, Yuan XQ, et al: Immune infiltration
and clinical outcome of super-enhancer-associated lncRNAs in
stomach adenocarcinoma. Front Oncol. 12:7804932022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Floros T and Tarhini AA: Anticancer
cytokines: Biology and clinical effects of interferon-α2,
interleukin (IL)-2, IL-15, IL-21, and IL-12. Semin Oncol.
42:539–548. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mao W, Wang K, Xu B, Zhang H, Sun S, Hu Q,
Zhang L, Liu C, Chen S, Wu J, et al: ciRS-7 is a prognostic
biomarker and potential gene therapy target for renal cell
carcinoma. Mol Cancer. 20:1422021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chan KK, Matchett KB, Coulter JA, Yuen HF,
McCrudden CM, Zhang SD, Irwin GW, Davidson MA, Rülicke T, Schober
S, et al: Erythropoietin drives breast cancer progression by
activation of its receptor EPOR. Oncotarget. 8:38251–38263. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tasian SK, Loh ML and Hunger SP:
Philadelphia chromosome-like acute lymphoblastic leukemia. Blood.
130:2064–2072. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fujisue Y, Nakagawa T, Takahara K, Inamoto
T, Kiyama S, Azuma H and Asahi M: Induction of erythropoietin
increases the cell proliferation rate in a hypoxia-inducible
factor-1-dependent and -independent manner in renal cell carcinoma
cell lines. Oncol Lett. 5:1765–1770. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gryko M, Pryczynicz A, Zareba K, Kędra B,
Kemona A and Guzińska-Ustymowicz K: The expression of Bcl-2 and BID
in gastric cancer cells. J Immunol Res. 2014:9532032014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rupnarain C, Dlamini Z, Naicker S and
Bhoola K: Colon cancer: Genomics and apoptotic events. Biol Chem.
385:449–464. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fan Y, Wang X and Li Y: IFI30 expression
predicts patient prognosis in breast cancer and dictates breast
cancer cells proliferation via regulating autophagy. Int J Med Sci.
18:3342–3352. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yuan Y, Qin H, Li H, Shi W, Bao L, Xu S,
Yin J and Zheng L: The functional roles of ISG15/ISGylation in
cancer. Molecules. 28:13372023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Qu T, Zhang W, Yan C, Ren D, Wang Y, Guo
Y, Guo Q, Wang J, Liu L, Han L, et al: ISG15 targets glycosylated
PD-L1 and promotes its degradation to enhance antitumor immune
effects in lung adenocarcinoma. J Transl Med. 21:3412023.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lu JF, Hu ZQ, Yang MX, Liu WY, Pan GF,
Ding JB, Liu JZ, Tang L, Hu B and Li HC: Downregulation of PDGF-D
inhibits proliferation and invasion in breast cancer MDA-MB-231
cells. Clin Breast Cancer. 22:e173–e183. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yanru W, Zhenyu B, Zhengchuan N, Qi Q,
Chunmin L and Weiqiang Y: Transcriptomic analyses of chemokines
reveal that down-regulation of XCR1 is associated with advanced
hepatocellular carcinoma. Biochem Biophys Res Commun.
496:1314–1321. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Han TD, Shang DH and Tian Y: Docetaxel
enhances apoptosis and G2/M cell cycle arrest by suppressing
mitogen-activated protein kinase signaling in human renal clear
cell carcinoma. Genet Mol Res. 15:2016. View Article : Google Scholar
|
|
39
|
Sumitomo M, Koizumi F, Asano T, Horiguchi
A, Ito K, Asano T, Kakizoe T, Hayakawa M and Matsumura Y: Novel
SN-38-incorporated polymeric micelle, NK012, strongly suppresses
renal cancer progression. Cancer Res. 68:1631–1635. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Motzer RJ, Hutson TE, Cella D, Reeves J,
Hawkins R, Guo J, Nathan P, Staehler M, de Souza P, Merchan JR, et
al: Pazopanib versus sunitinib in metastatic renal-cell carcinoma.
N Engl J Med. 369:722–731. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
El-Galley R, Keane TE and Sun C:
Camptothecin analogues and vinblastine in the treatment of renal
cell carcinoma: An in vivo study using a human orthotopic renal
cancer xenograft. Urol Oncol. 21:49–57. 2003. View Article : Google Scholar : PubMed/NCBI
|