Introduction
Lung cancer (LC) is the leading cause of
cancer-related mortality and morbidity, with 1.8 million deaths
accounting for 18% of the global cancer-associated mortality rate
(1,2). The estimated 5-year survival rate of
patients with LC is 68–92% when diagnosed at an early stage.
However, if detected at late stages, this drops to 10% (3). Patients with LC have a poor prognosis
because the disease is frequently detected at advanced stages
without curative treatment options. One of the key reasons for such
a poor prognosis is the lack of efficient diagnostic tools for
early detection (4).
Screening with low-dose computed tomography (CT)
scans of the chest has been introduced as a potential tool for
early detection of LC (5). Although
low-dose CT allows the detection of early-stage LC with a high
sensitivity, which can reduce LC-associated mortality, it has
numerous drawbacks including radiation exposure, a high false
positive rate and a low diagnostic accuracy (6–8).
Furthermore, LC screening by chest x-ray and sputum cytology has
failed to overcome early detection and risk assessment limitations,
thus failing to improve overall survival (9). Because there is currently no early
detection method in clinical practice, a patient with suspected LC
is typically subjected to a full clinical workup that includes CT
scanning followed by bronchoscopy (7).
Bronchoscopy is the most common invasive procedure
used to diagnose LC. However, the diagnostic yield of bronchoscopy
is unsatisfactory, with ambiguous results in half of patients
suspected of having LC, especially for peripheral tumors (10,11).
If bronchoscopy fails to detect LC, further invasive diagnostic
procedures, such as transthoracic needle aspiration or surgical
lung biopsy, may be required, posing a significant risk of
complications, such as pneumothorax, bleeding and infection
(12). Bronchoscopy is the
preferred method for confirming suspected lung lesions through
pathological assessment of a tissue biopsy or cytological specimen
obtained during bronchoscopy (11,13).
Cytology examinations include bronchial brushing, bronchial washing
(BW) and bronchoalveolar lavage samples (14). Cytology often results in an
equivocal or inconclusive result, even when performed by
experienced professionals. In addition, the sensitivity range of
cytology is low (23.5–32.1%) (15).
Nevertheless, cytological sampling is a minimally invasive, safe
and well-tolerated method performed during bronchoscopy for LC
diagnosis (10,11,14).
Biomarkers may also be used as a diagnostic adjunct to resolve
equivocal cytology results (16).
Therefore, development of molecular biomarker tests with higher
sensitivity for routine cytology specimens represents an effective
approach to improving the diagnostic yield of bronchoscopy
(17).
DNA methylation, a key epigenetic phenomenon, plays
a fundamental role in various biological processes, including
development, cell differentiation, aging, tumorigenesis and other
disease (18). Abnormal DNA
methylation is involved in tumor development; it is one of the
earliest and most frequent genomic alterations during
carcinogenesis (9,19). The analysis of DNA methylation
biomarkers provides potential for early detection of LC (10,14,20,21). A
number of genes, such as adenomatous polyposis coli,
ras-association (RalGDS/AF-6) domain family member 1
(RASSF1A), p16, short-stature homeobox 2
(SHOX2) and various homeobox genes, have been extensively
investigated to diagnose LC (22,23).
Nonetheless, no established biomarker test with clinical
application for LC detection exists.
In the present study, CpG methylation microarray
analysis was performed to investigate a subset of differentially
hypermethylated genes in primary lung tumors compared with paired
adjacent non-tumor tissues. The aim of the study was to identify
methylation biomarkers for the early detection of LC and to
validate them clinically using BW samples.
Materials and methods
Cell lines and clinical samples
The human LC cell lines A549 (cat. no. CCL-185),
NCI-H358 (cat. no. CRL-5807), SK-MES-1 (cat. no. HTB-58) and
NCI-H146 (cat. no. HTB-173) were obtained from the American Type
Culture Collection and cultured in RPMI-1640 (cat. no. LM011-06;
Welgene, Inc.) in a humidified 5% CO2 incubator at 37°C,
supplemented with 10% fetal bovine serum (cat. no. S001-04,
Welgene, Inc.). Normal human bronchial epithelial (NHBE) cells were
purchased from Cambrex Bio Science Rockland, Ltd. (cat. no. CC2540)
and cultured in BEBM (cat. no. cc-3171; Cambrex Bio Science
Rockland, Ltd.) in a humidified 5% CO2 incubator at
37°C. The cultured cells were tested for mycoplasma every month to
ensure that they were not contaminated using a MycoStrip kit (cat.
no. rep-mys-20; InvivoGen). Cells were authenticated by short
tandem repeat analysis using GenePrint® 10 System
(cat. no. B9510; Promega Corporation).
Fresh-frozen primary tumors and paired adjacent
non-tumor tissue from 13 patients with non-small cell LC (NSCLC) at
various stages (I, n=5; II, n=5; III, n=3) were obtained from the
Biobank of Chungnam National University Hospital (Daejeon, South
Korea), which participates in the Korea Biobank Project (KBP).
Tissue specimens were collected at the time of surgery between
February 2014 and December 2019. Every tumor specimen was
histologically verified by a board-certified pathologist.
All BW samples were provided by Konyang University
Hospital (Daejeon, South Korea). All individuals donating BW
samples for the present study were investigated for suspected LC.
All BW samples were collected during flexible fiberoptic
bronchoscopy (Olympus Corporation) by aspiration with a flexible
bronchoscope from the region of the suspicious lesion between April
2022 and March 2023. Cytological diagnostics was performed by a
board-certified pathologist. Briefly, 5–10 ml sterile normal saline
was instilled two or three times. Fluid (≥10 ml) was then retrieved
into a preservative buffer (Genomictree, Inc.). A total of 101 BW
samples were obtained from 68 patients with LC (49 NSCLC and 19
SCLC) and 33 individuals with benign diseases and were used for
methylation analysis. The patient clinicopathological and
demographical information is shown in Table I.
 | Table I.Clinicopathological features of
tissue and bronchial washing samples. |
Table I.
Clinicopathological features of
tissue and bronchial washing samples.
| Characteristic | Tissue | Bronchial
washing |
|---|
| Sex (%) |
|
|
| Non-LC
(benign) | - | 33 (100.0) |
|
Male | - | 16 (48.5) |
|
Female | - | 17 (51.5) |
| LC | 13 | 68 (100.0) |
|
Male | 12 (77.8) | 53 (77.9) |
|
Female | 1 (22.2) | 15 (22.1) |
| Mean age (range),
years |
|
|
| Non-LC
(benign) | - | 67.0 (47–87) |
| LC | 52.4 (58–81) | 71.7 (49–93) |
| Pathological stage
(%) |
|
|
| I | 5 (38.5) | 13 (19.1) |
| II | 5 (38.5) | 6 (8.8) |
|
III | 3 (23.1) | 14 (20.6) |
| IV | - | 35 (51.5) |
| Histology (%) |
|
|
|
NSCLC |
| 49 (72.1) |
|
ADC | 6 (46.2) | 27 (39.7) |
|
SCC | 7 (53.8) | 20 (29.4) |
|
Othera | - | 2 (2.9) |
|
SCLC | - | 19 (27.9) |
| Tumor location
(%) |
|
|
|
Central | - | 34 (50.0) |
|
Peripheral | - | 34 (50.0) |
The present study adhered to local ethics guidelines
and was approved by the Institutional Review Board of the Chungnam
National University Hospital (approval no. 2022-02-061-002, Dajeon,
South Korea) and Konyang University Hospital (approval no.
2022-03-025, Dajeon, South Korea). Written informed consent was
obtained from all patients.
CpG methylation microarray
analysis
To identify differentially methylated genes in
primary lung tumors and paired adjacent non-tumor tissues, CpG
methylation microarray analyses were conducted using 0.5 µg genomic
DNA isolated from 13 patients with LC. CpG methylation microarray
analysis was performed as described previously (24) using human CpG island microarray kit,
244k (Agilent Technologies, Inc.) according to the manufacturer's
instructions. Raw CpG methylation microarray data were submitted to
Gene Expression Omnibus (accession no. GSE246510;
ncbi.nlm.nih.gov/geo). The hybridized images were analyzed and
quantified using Agilent Feature Extraction (version 9.3.2.1;
Agilent Technologies, Inc.) and GeneSpring (version 7.3.1; Agilent
Technologies, Inc.).
To determine differentially hypermethylated
candidate genes in primary tumor compared with paired adjacent
non-tumor tissue samples, statistical analysis was performed using
a parametric ANOVA test with Benjamini and Hochberg multiple
testing correction (P<0.05), followed by fold-change analysis.
Mean fold change was calculated by dividing mean methylation levels
in tumor tissue by mean methylation levels in non-tumor tissue.
Multiple-probe enriched genes were selected as methylation
candidate genes if their probes yielded a positive call for
methylation in the lung primary tumor compared with non-tumor
tissue with at least two adjacent probes, allowing for a one-gap
probe within CpG islands.
DNA isolation and bisulfite
treatment
Genomic DNA was isolated from cell lines and tissues
using the QIAmp DNA Mini kit (cat. no. 51304; Qiagen GmbH)
according to the manufacturer's instructions. Genomic DNA was
isolated from BW samples using a solid phase magnetic bead-based GT
NUCLEIC ACID PREP kit (cat. no. GT-PREP-1; Genomictree, Inc.)
according to the manufacturer's instructions. Genomic DNA was
chemically modified with sodium bisulfite using an EZ DNA
Methylation Gold kit (cat. no. D5006; Zymo Research Corp.)
according to the manufacturer's instructions. Bisulfite-converted
DNA was purified and eluted with an elution buffer using a
Zymo-Spin IC column (Zymo Research Corp.).
Methylation analysis by
bisulfite-pyrosequencing
Candidate methylation targets were analyzed for
methylation levels using bisulfite-pyrosequencing, as previously
described, with slight modifications (24). Bisulfite-treated genomic DNA
underwent PCR amplification targeting the region of interest,
employing a specific primer set. Either the forward or reverse
primer was biotinylated, facilitating generation of a
single-stranded DNA template for subsequent pyrosequencing on a
PyroMark Q48 Autoprep (Qiagen GmbH). Pyrosequencing primers were
designed to detect methylated cytosine by analyzing CpG
dinucleotide sites within the target sequences of bisulfite-treated
DNA. To facilitate primer design, nucleotide sequences of the
candidate genes were retrieved from the NCBI Reference Sequence
database (ncbi.nlm.nih.gov/refseq/) and converted into
bisulfite-treated sequences. Both PCR primers and sequencing
primers were designed to complementarily bind to the
bisulfite-converted sequences within the regions of the interest,
while avoiding CpGs within the primer sequences, using PyroMark
Assay Design Software V 2.0 (Qiagen GmbH). Primer sequences are
listed in Table SI. Briefly, 20 ng
bisulfite-modified DNA was amplified in a 20 µl reaction with a
gene-specific primer set and TOPsimple PCR DryMix-HOT (cat. no.
P581H; Enzynomics, Inc.). PCR amplification was conducted under the
following thermocycling conditions: 95°C for 10 min, followed by 40
cycles of 95°C for 45 sec, optimal annealing temperature for 45
sec, and 70°C for 45 sec for each target. Pyrosequencing was
performed using a PyroMark Gold Q48 reagent cartridge and a
PyroMark Q48 instrument (Qiagen GmbH) according to the
manufacturer's instructions.
The methylation index (MtI) of every gene in every
sample was calculated as the mean value of methylated
cytosine/(methylated cytosine + unmethylated cytosine) for all
examined CpGs in target regions. All pyrosequencing reactions
included a negative control without DNA template.
Methylation-positive was considered if the MtI of the primary tumor
was greater than that of the corresponding non-tumor tissue.
Assessment of methylation status in BW
samples using linear target enrichment (LTE)-quantitative
methylation-specific PCR (qMSP) assay
A 3-plex LTE-qMSP assay was developed, integrating
two methylation targets from candidate genes, and the control gene
collagen type II α1 (COL2A1) within a closed single-tube
system to assess the methylation status of candidate targets in
BW-derived DNA samples. This method involves two sequential rounds
of PCR. LTE first employs one-direction PCR targeting to linearly
enrich DNA of methylation candidates, followed by qMSP for
exponential amplification of both the methylated target and the
control gene.
In the first round of PCR, high annealing
temperature (70°C) was applied to facilitate unidirectional DNA
synthesis while preventing other primers with regular melting
temperatures from initiating DNA synthesis. Two specific primers
with a universal tag sequence (UTS) at the 5′ end were designed to
anneal to two distinct methylation target genes. The template DNA
synthesis occurred in one direction only, replicating with each
cycle of PCR.
In the subsequent PCR, the reaction was conducted at
a reduced temperature (60°C). A total of three sets of primers and
probes were utilized targeting amplification of the two methylation
targets and the control gene. The forward primers for the
methylation targets were designed to bind specifically to the
respective methylation target sites and UTS was used as reverse
primer. The control target utilized a primer set specific to a DNA
region of COL2A1 gene lacking CpG dinucleotides, with
annealing at 60°C. Probes were designed to bind internal sites of
each PCR product, thereby generating signals indicated of PCR
product formation.
To design primers and probes for LTE-qMSP,
nucleotide sequences corresponding to the genes of interest were
obtained from the NCBI Reference Sequence database
(ncbi.nlm.nih.gov/refseq/). The primer and probe sequences were
designed to bind complementarily to the bisulfite-converted
sequences of the methylation target regions using MethPrimer
program (version 2.0;
urogene.org/cgi-bin/methprimer/methprimer.cgi). The primer and
probe sequences are provided in Table
SII.
For every reaction, 20 ng BW-derived DNA underwent
bisulfite conversion. The bisulfite-converted DNA was purified and
eluted with 12 µl elution buffer, serving as input for the LTE-qMSP
assay. The reaction mixture (25 µl) comprised 10 µl input DNA and
15 µl reagent containing two methylation target
methylation-specific reverse primer with a 5′ UTS, two
methylation-specific forward primers, UTS as reverse primer, two
probes for specific methylation sites of targets,
COL2A1-specific forward and reverse primers, COL2A1
probe and 5 µl of the master mix, TOPreal™ Fast qPCR 5X PreMIX
TaqMan-Probe (Enzynomics, Inc.). LTE-qMSP reaction was performed on
an AB7500 FAST Real-Time PCR system (Thermo Fisher Scientific,
Inc.) under the following thermocycling conditions: 95°C for 5 min,
5 cycles of 95°C for 15 sec and 70°C for 45 sec, followed by 35
cycles of 95°C for 15 sec and 60°C for 45 sec.
The relative methylation in each sample was
calculated as 35-ΔCT [CT of the amplified
target gene-CT of COL2A1 (human reference gene)]
(25). A higher value indicates a
greater level of methylation. If the CT of the target
gene was undetectable, the value was set to 25, the closest value
to the lowest 35-ΔCT among all test results. The assay
was conducted by trained personnel blinded to the bronchoscopy or
the histopathology results.
Statistical analysis
All statistical analyses were performed using
MedCalc (version 9.3.2.0, medcalc.org/). Receiver operating
characteristic (ROC) curves were constructed to evaluate the test
performance, with calculation of the area under ROC (AUC) and 95%
confidence intervals (CIs). Methylation tests were performed once
for each sample, and the data are presented as the mean and
standard deviations of all test results for the samples. P<0.05
was considered to indicate a statistically significant
difference.
To calculate sensitivity and specificity in BW
samples, test results were categorized as follows:
Methylation-positive as ‘1’ and methylation-negative as ‘0’. A
binary logistic regression analysis was used to determine the best
performing marker combination for detecting LC in BW samples. To
describe demographic and other clinical characteristics, frequency
and percent were used. The negative predictive value (NPV) and
positive predictive value (PPV) were also calculated.
The paired t test was conducted to compare
differences in methylation levels of each gene between tumor and
paired adjacent non-tumor tissue. The Kruskal Wallis test was used
to analyze differences in methylation levels between patients with
LC and individuals with benign diseases in BW samples. Spearman's
correlation analysis was performed to investigate the correlation
between methylation levels of genes in BW specimens. The difference
in the sensitivity and specificity between the methylation testing
and cytology examination for LC diagnosis was analyzed using
McNemar test. Fisher's exact test was utilized to examine the
relationship between clinicopathological parameters and methylation
status in BW samples.
Results
Identification of candidate genes
hypermethylated in primary lung tumor tissue
To investigate a subset of hypermethylated candidate
genes for detecting LC, CpG methylation patterns were compared
between primary lung tumors and corresponding adjacent non-tumor
tissues using CpG methylation microarray analyses (Fig. S1). The initial hypothesis was that
candidate genes should be unmethylated in non-tumor tissues and
frequently hypermethylated in tumor tissues. A total of 18,585
unmethylated CpG probes across all 13 non-tumor tissues were
selected. Subsequent statistical analysis (ANOVA) identified 2,844
CpG probes differentially hypermethylated in primary tumor tissue.
Methylation candidate numbers were then narrowed to 516 CpG probes
representing 65 annotated genes that showed consistent
hypermethylation in at least two adjacent CpG probes (Table SIII). Among the 65 candidate genes,
the analysis focused on 10 hypermethylated genes, apoptosis
antagonizing transcription factor, ATP-binding cassette subfamily C
member 9, ADAM metallopeptidase with thrombospondin type 1 motif 20
(ADAMTS20), forkhead box C2 (mesenchyme forkhead 1)
(FOXC2), hey-like transcriptional repressor, NK2
transcription factor-related locus 5 (Drosophila)
(NKX2-5), oligodendrocyte transcription factor 3
(OLIG3), one cut domain family member 1, protocadherin γ
subfamily A 12 (PCDHGA12) and paired-related homeobox 1
(PRRX1), exhibiting a positive call for methylation in their
5′ regulatory regions (promoter or 5′ untranslated region) and had
not been previously reported as aberrantly hypermethylated in
primary lung tumor tissue.
Verification of methylation candidate
genes in LC cell lines and tissues using
bisulfite-pyrosequencing
To verify the methylation status of 10 candidate
genes, a pyrosequencing-based methylation assessment was performed
in four representative LC cell lines, A549, NCI-H358, SK-MES-1 and
NCI-H146, and their status was compared with that of NHBE cells.
Results revealed that all 10 genes were hypermethylated in LC cell
lines but methylated at a low level in NHBE cells (Fig. 1). In verification analysis using a
pyrosequencing assay to confirm whether these candidate genes were
aberrantly hypermethylated in primary lung tumors examined in CpG
microarray analysis, all candidate genes except HELT
exhibited a significantly high level of methylation in primary
tumor tissues compared with their corresponding non-tumor tissues
(Fig. 2). The mean MtIs of all
candidate genes were high in lung tumors (range, 25.1–60.4%), but
relatively low in non-tumor tissues (range, 5.6–40.7%; Table II). Among these genes, six
(ADAMTS20, FOXC2, NKX2-5, OLIG3, PCDHGA12 and PRRX1)
were chosen for further validation because of their consistently
high (100%) methylation positivity across all tumor tissues.
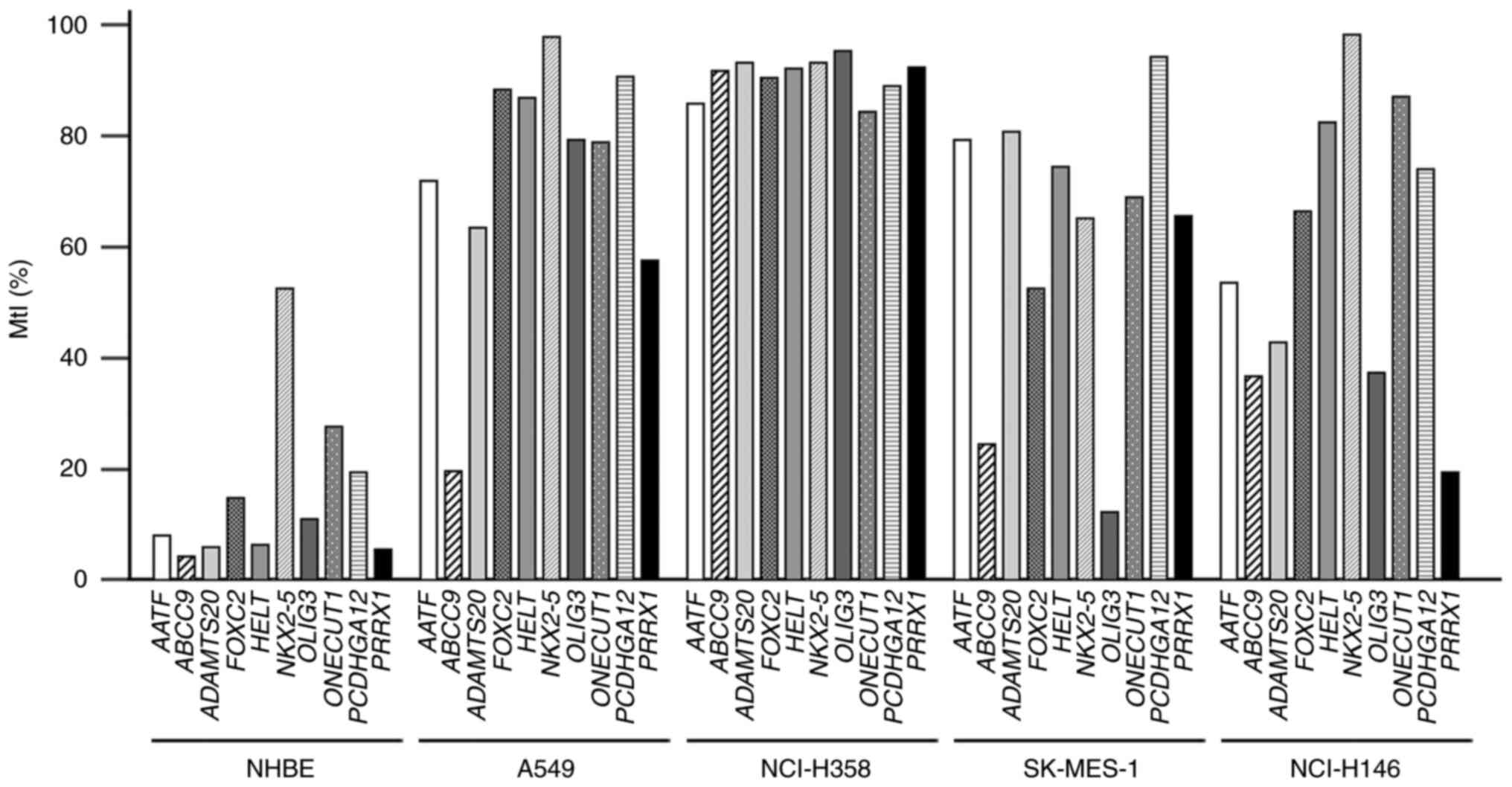 | Figure 1.Bisulfite-pyrosequencing results of
10 candidate genes in cell lines. The methylation levels were
calculated for all examined CpG dinucleotides in target regions.
MtI values for each gene were plotted in four LC cell lines A549,
NCI-H358, SK-MES-1 and NCI-H146 and in NHBE cells. MtI, methylation
index; NHBE, normal human bronchial epithelial cell; AATF,
apoptosis antagonizing transcription factor; ABCC9,
ATP-binding cassette subfamily C member 9; ADAMTS20, ADAM
metallopeptidase with thrombospondin type 1 motif 20; FOXC2,
forkhead box C2 (mesenchyme forkhead 1); HELT, hey-like
transcriptional repressor; NKX2-5, NK2 transcription
factor-related, locus 5 (Drosophila); OLIG3,
oligodendrocyte transcription factor 3; ONECUT1, one cut
domain family member 1; PCDHGA12, protocadherin γ subfamily
A, 12; PRRX1, paired-related homeobox 1. |
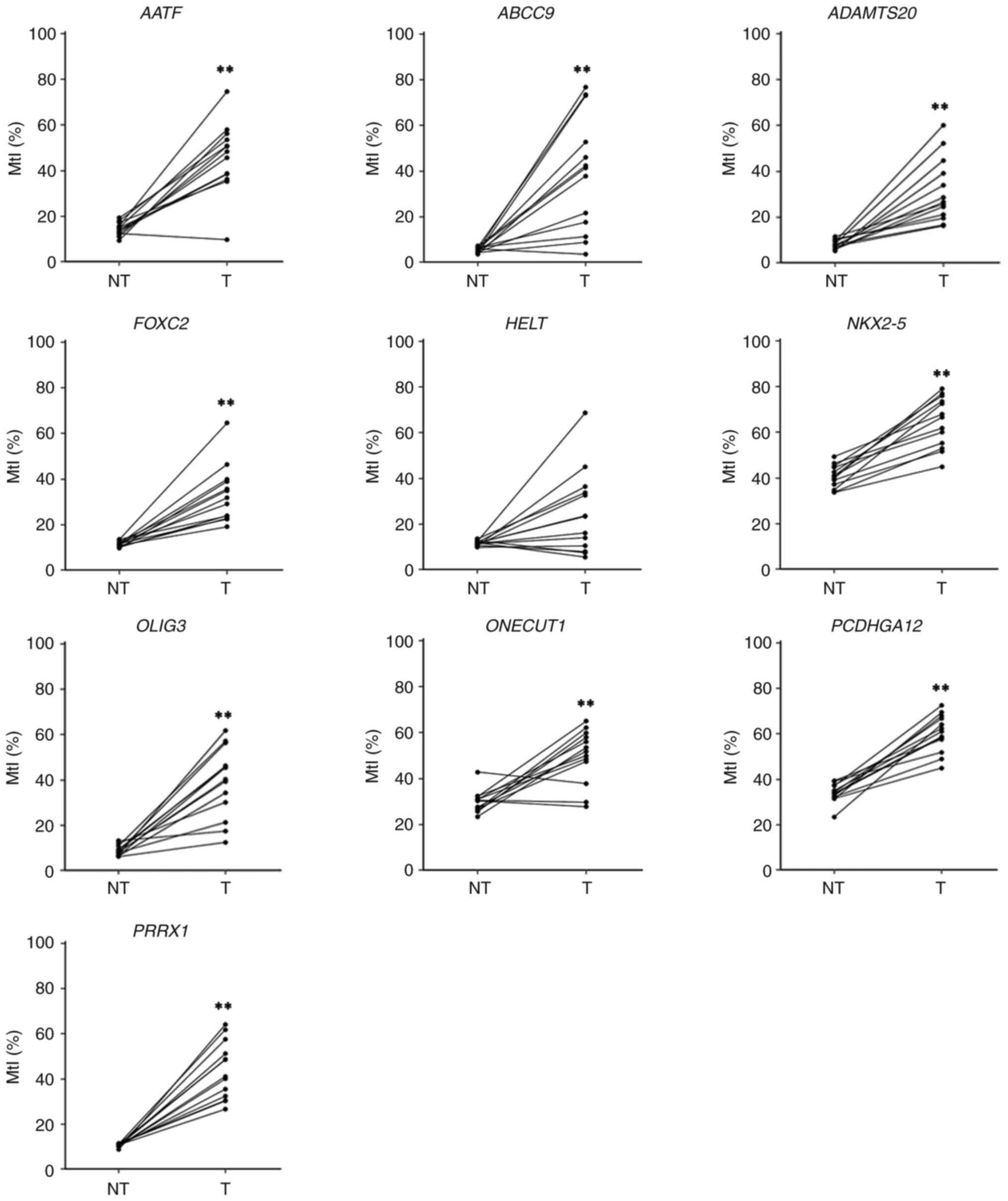 | Figure 2.Assessment of methylation levels of
10 candidate genes in paired lung tissue by
bisulfite-pyrosequencing. MtI for each gene was determined in
primary lung tumor and paired adjacent NT tissue used for the CpG
methylation microarray analysis. Samples from the same patients are
linked with a straight line. **P<0.01 vs. NT. NT, non-tumor, T,
tumor. MtI, methylation index; NHBE, normal human bronchial
epithelial cell; AATF, apoptosis antagonizing transcription
factor; ABCC9, ATP-binding cassette subfamily C member 9;
ADAMTS20, ADAM metallopeptidase with thrombospondin type 1
motif 20; FOXC2, forkhead box C2 (MFH-1, mesenchyme forkhead
1); HELT, Hey-like transcriptional repressor; NKX2-5,
NK2 transcription factor-related, locus 5 (Drosophila);
OLIG3, oligodendrocyte transcription factor 3;
ONECUT1, one cut domain family member 1; PCDHGA12,
protocadherin γ subfamily A, 12; PRRX1, paired-related
homeobox 1. |
| Table II.Methylation status of 10 candidate
genes in 13 paired lung tissues used in CpG methylation microarray
analysis. |
Table II.
Methylation status of 10 candidate
genes in 13 paired lung tissues used in CpG methylation microarray
analysis.
|
| Mean MtI, % |
|
|
|---|
|
|
|
|
|
|---|
| Gene | Paired adjacent
non-tumor tissue | Primary tumor
tissue | Methylation
positivity, % (positive samples/total samples) | P-value |
|---|
| AATF | 14.4±2.8 | 45.9±15.3 | 92.3 (12/13) | <0.001 |
| ABCC9 | 5.6±1.2 | 39.0±25.3 | 92.3 (12/13) | <0.001 |
|
ADAMTS20 | 8.0±1.9 | 31.5±13.9 | 100 (13/13) | <0.001 |
| FOXC2 | 11.5±1.2 | 33.4±12.5 | 100 (13/13) | <0.001 |
| HELT | 11.6±1.0 | 25.1±18.2 | 76.9 (10/13) | 0.081 |
| NKX2-5 | 40.7±4.9 | 64.6±11.0 | 100 (13/13) | <0.001 |
| OLIG3 | 8.7±2.2 | 39.1±15.5 | 100 (13/13) | <0.001 |
| ONECUT1 | 29.8±4.8 | 49.8±11.7 | 76.9 (10/13) | <0.001 |
|
PCDHGA12 | 34.0±4.2 | 60.4±8.2 | 100 (13/13) | <0.001 |
| PRRX1 | 10.6±0.7 | 43.8±12.6 | 100 (13/13) | <0.001 |
Clinical validation of six methylated
genes for detecting LC using BW specimens
A highly sensitive and accurate 3-plex LTE-qMSP in a
single closed tube was developed to measure the methylation of
target genes in BW samples. Methylation levels were determined
using DNA from 68 patients with LC and 33 individuals with benign
diseases. Results of the 3-plex LTE-qMSP showed that the
methylation levels of all six genes were significantly higher in
patients with LC than in individuals with benign diseases (Fig. 3). To determine sensitivity and
specificity of individual genes for LC detection, ROC analysis was
performed (Fig. 4). Given the
optimal cut-off values, the sensitivity range of each gene was
52.9–80.9% and the specificity range was 81.8–97.0% for LC; AUC
range was 0.696–0.859. The PPV range was 86.7–97.9% and NPV range
was 48.2–69.8%. Cytology results of BW specimens showed a
sensitivity of 50.0%, with a specificity of 100% (Table III).
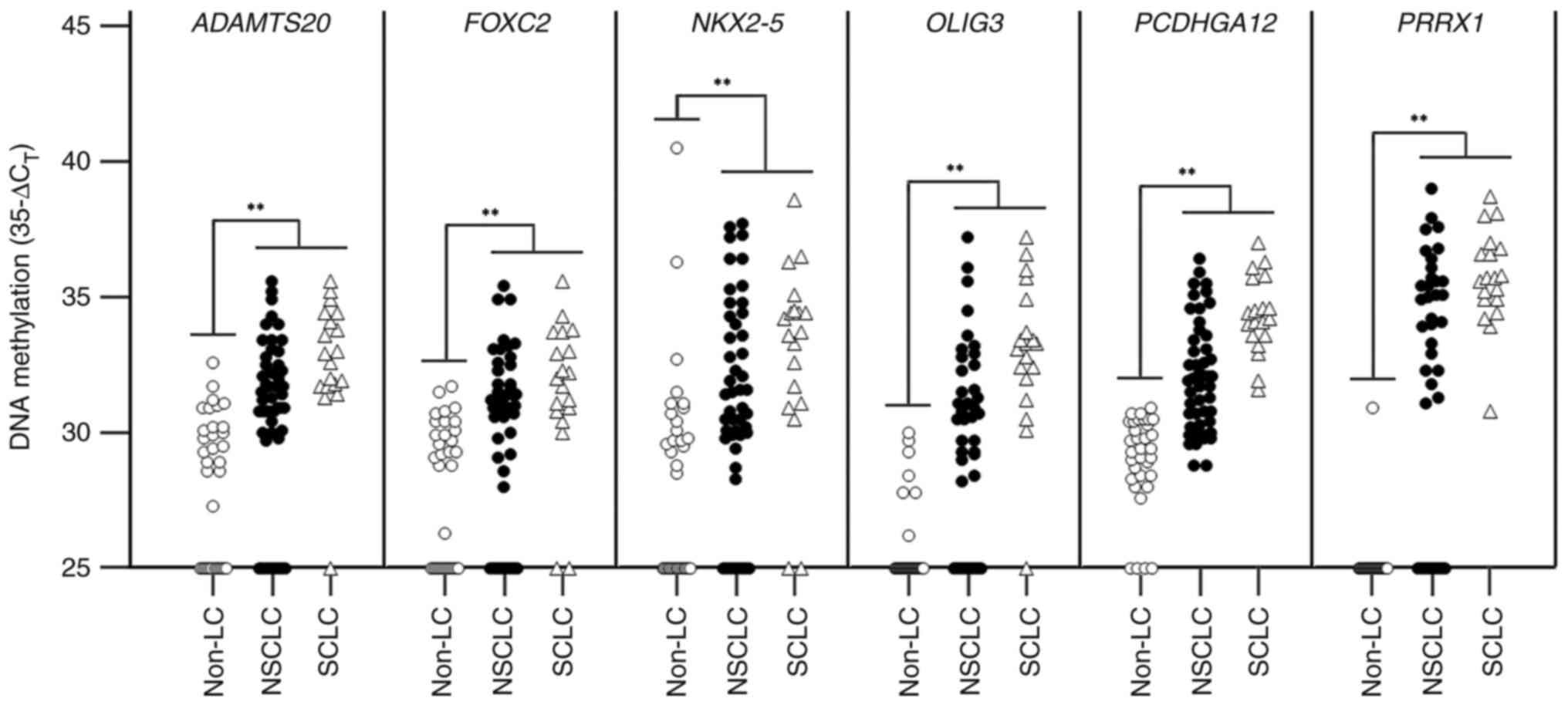 | Figure 3.Clinical validation of six genes in
bronchial washing samples using linear target
enrichment-quantitative methylation-specific PCR. The distribution
of methylation for every gene is depicted as scatter plots of
35-ΔCT value. The differences in methylation levels
between patients with lung cancer and individuals with benign
diseases are statistically compared. **P<0.01. Non-LC,
individuals with benign diseases, NSCLC, non-small cell lung
cancer; ADAMTS20, ADAM metallopeptidase with thrombospondin
type 1 motif 20; FOXC2, forkhead box C2 (mesenchyme forkhead
1); NKX2-5, NK2 transcription factor-related, locus 5
(Drosophila); OLIG3, oligodendrocyte transcription
factor 3; PCDHGA12, protocadherin γ subfamily A, 12;
PRRX1, paired-related homeobox 1. |
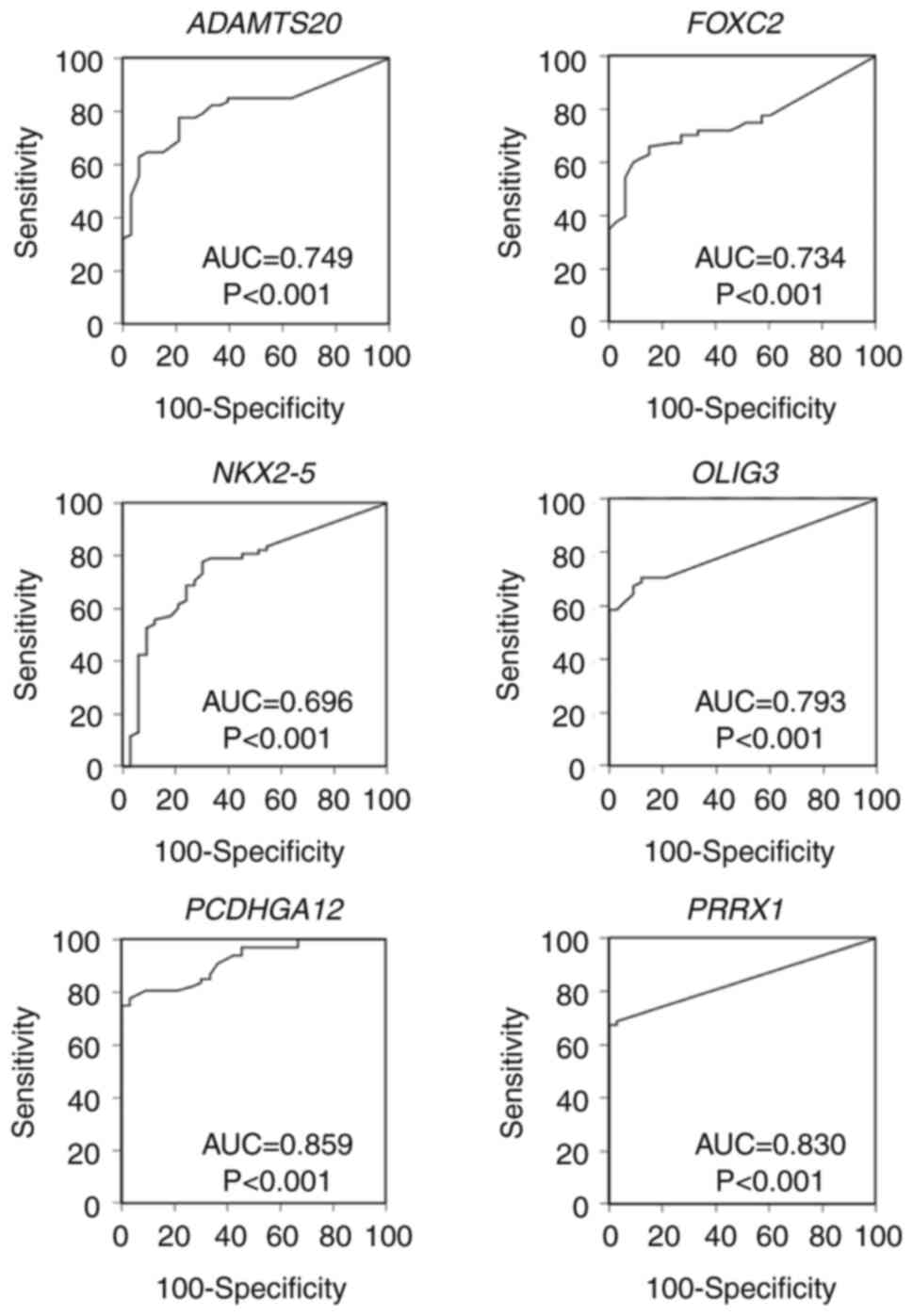 | Figure 4.ROC analysis of six genes using BW
samples. ROC curves for six genes were analyzed for differentiating
LC from non-LC using BW samples. ROC, receiver operating
characteristic; BW, bronchial washing; LC, lung cancer; AUC, area
under ROC; ADAMTS20, ADAM metallopeptidase with
thrombospondin type 1 motif 20; FOXC2, forkhead box C2
(mesenchyme forkhead 1); NKX2-5, NK2 transcription
factor-related, locus 5 (Drosophila); OLIG3,
oligodendrocyte transcription factor 3; PCDHGA12,
protocadherin γ subfamily A, 12; PRRX1, paired-related
homeobox 1. |
| Table III.Clinical performance of 6 genes and
cytology in detecting lung cancer using bronchial washing
samples. |
Table III.
Clinical performance of 6 genes and
cytology in detecting lung cancer using bronchial washing
samples.
| Test | Cut-off
(35-ΔCT) | AUC (95% CI) | Sensitivity, % (95%
CI) | Specificity, % (95%
CI) | PPV, % (95%
CI) | NPV, % (95%
CI) |
|---|
|
ADAMTS20 | 31.5 | 0.749
(0.653–0.830) | 55.9
(43.3–67.9) | 94.0
(79.8–99.3) | 95.0
(83.0–98.7) | 50.8
(43.8–57.8) |
| FOXC2 | 31.0 | 0.734
(0.637–0.817) | 52.9
(40.4–65.2) | 93.9
(79.8–99.3) | 94.7
(82.2–98.6) | 49.2
(42.6–55.8) |
| NKX2-5 | 31.0 | 0.696
(0.596–0.783) | 57.4
(44.8–69.3) | 81.8
(64.5–93.0) | 86.7
(75.4–93.2) | 48.2
(40.4–56.2) |
| OLIG3 | 28.5 | 0.793
(0.701–0.867) | 67.6
(55.2–78.5) | 90.9
(75.7–98.1) | 93.9
(93.7–97.9) | 57.7
(48.7–66.2) |
|
PCDHGA12 | 30.5 | 0.859
(0.776–0.920) | 80.9
(69.5–89.4) | 90.9
(75.7–98.1) | 94.8
(86.1–98.2) | 69.8
(58.3–79.2) |
| PRRX1 | 25.0 | 0.830
(0.743–0.898) | 69.1
(56.7–79.8) | 97.0
(84.2–99.9) | 97.9
(87.1–99.7) | 60.4
(51.5–68.6) |
| Cytology | - | 0.750
(0.654–0.831) | 50.0
(37.6–62.4) | 100.0
(89.4–100.0) | 100.0 | 49.3
(43.4–55.2) |
| PCDHGA12 or
PRRX1 | - | 0.891
(0.813–0.944) | 82.4
(71.2–90.2) | 87.9
(71.8–96.6) | 93.3
(84.7–97.2) | 70.7
(58.7–80.4) |
| PCDHGA12 or
PRRX1 or cytology | - | 0.866
(0.784–0.926) | 85.3
(74.6–92.7) | 87.9
(71.8–96.6) | 93.6
(85.2–97.3) | 74.4
(61.7–83.9) |
In single marker analysis, PCDHGA12 showed
the highest sensitivity of 80.9% (55/68 patients with LC) and
PRRX1 achieved the highest specificity of 97.0% (32/33
individuals with benign diseases). PCDHGA12 and PRRX1
had relatively high diagnostic accuracies with AUCs of 0.859 and
0.830, respectively (Table III).
A binary logistic regression analysis was conducted using all six
markers to identify the best-performing biomarker combination in 36
splits (6 genes by 6 genes) of the dataset. PCDHGA12 and
PRRX1 biomarkers were significantly associated with LC.
These genes were highly co-methylated according to the correlation
analysis (Spearman's correlation coefficient, 0.74). The overall
sensitivity of the two-marker combination was 82.4% (56/68 patients
with LC; 95% CI, 71.2–90.2%) with a specificity of 87.9% (29/33
individuals with benign diseases; 95% CI, 71.8–96.6%) and AUC of
0.891 (95% CI, 0.813–0.944; Table
III). PPV was 93.3% (95% CI, 84.7–97.2%) and NPV was 70.7% (95%
CI, 58.7–80.4%). The sensitivities for stage I, II, III and IV LC
were 61.5% (8/13), 83.3% (5/6), 92.9% (13/14) and 85.7% (30/35),
respectively (Table IV). For
detecting LC, sensitivity of this two-marker combination
outperformed cytology (McNemar test). When combined with cytology
results, its sensitivity was slightly increased to 85.3% (58/68
patients with LC; 95% CI, 74.6–92.7%), maintaining specificity,
showing an AUC of 0.866 (95% CI, 0.784–0.926). PPV was 93.6% (95%
CI, 85.2–97.3%) and the NPV was 74.4% (95% CI, 61.7–83.9%; Table III).
| Table IV.Association between
clinicopathological parameters, methylation of two-marker model,
and cytology in bronchial washing specimens from 68 patients with
LC. |
Table IV.
Association between
clinicopathological parameters, methylation of two-marker model,
and cytology in bronchial washing specimens from 68 patients with
LC.
|
|
|
Methylation-positive (%) |
|---|
|
|
|
|
|---|
| Parameter | Samples | Two-marker | Cytology | Combined |
|---|
| Sex |
|
|
|
|
|
Male | 53 | 46 (86.8) | 28 (52.8) | 48 (90.6) |
|
Female | 15 | 10 (66.7) | 6 (40.0) | 10 (66.7) |
| P-value |
| 0.119 | 0.560 | 0.035 |
| Age, years |
|
|
|
|
|
<65 | 11 | 9 (81.8) | 6 (54.5) | 10 (90.9) |
|
≥65 | 57 | 47 (82.5) | 28 (49.1) | 48 (84.8) |
| P-value |
| >0.999 | >0.999 | >0.999 |
| Tumor location |
|
|
|
|
|
Central | 34 | 33 (97.1) | 21 (61.8) | 33 (97.1) |
|
Peripheral | 34 | 23 (67.6) | 13 (38.2) | 25 (73.5) |
| P-value |
| 0.003 | 0.089 | 0.013 |
| Histology |
|
|
|
|
|
NSCLC | 49 | 37 (75.5) | 24 (49.0) | 39 (79.6) |
|
SCLC | 19 | 19 (100.0) | 10 (52.6) | 19 (100.0) |
| P-value |
| 0.015 | 0.795 | 0.052 |
| Stage |
|
|
|
|
| I | 13 | 8 (61.5) | 1 (7.7) | 8 (61.5) |
| II | 6 | 5 (83.3) | 4 (66.7) | 5 (83.3) |
|
III | 14 | 13 (92.9) | 8 (57.1) | 13 (92.9) |
| IV | 35 | 30 (85.7) | 21 (60.0) | 32 (91.4) |
|
P-valuea |
| 0.080 | 0.029 | 0.027 |
Subgroup analysis revealed that the methylation
status of the two-marker combination was not associated with sex,
age or stage (Fisher's exact test). However, it was associated with
tumor location and histology (Fisher's exact test) (Table IV). Notably, the sensitivity for
SCLC was 100% (19/19 patients with SCLC; Table IV). Specificity was not
significantly affected by sex or age (Fisher's exact test).
Discussion
To the best of our knowledge, the present study is
the first to demonstrate that the novel combination of methylation
biomarkers PCDHGA12 and PRRX1 effectively detects
early-stage LC. This test may identify patients with nodules who
may not require invasive procedures. A large-scale prospective
clinical study is warranted to validate its performance for
clinical use.
For diagnosing LC, cytology examinations using BW
specimens remain a preferred method due to their minimally invasive
nature and safety profile during bronchoscopy, but often yield
inconclusive results, even when performed by experienced
professionals (11,14,15).
Despite widespread use, the sensitivity of cytology remains
relatively low, with a range of 23.5–32.1% (15). Specific biomarkers for LC are
promising diagnostic adjuncts to confirm equivocal cytology
findings. Development of molecular biomarker tests using BW
specimens presents a promising strategy for enhancing diagnostic
accuracy of bronchoscopy (16,17).
Aberrant DNA methylation is considered one of the
most influential epigenetic biomarkers in various types of cancer,
including LC (9,10,14).
In the present study, a genome-wide search using CpG methylation
microarray analysis was conducted to identify genes hypermethylated
in LC. A total of 10 candidate genes consistently hypermethylated
in primary lung tumors compared with paired adjacent non-tumor
tissues were selected. A stepwise validation process using
bisulfite-pyrosequencing assay identified six potential methylation
biomarker candidates for diagnosing LC. Subsequent verification of
those biomarkers using LTE-qMSP assay demonstrated that two
methylation biomarkers, PCDHGA12 and PRRX1, had a
high potential for detecting LC in BW specimens as a diagnostic
adjunct to cytology.
Several studies have reported multiple methylation
biomarkers for diagnosing LC using BW specimens (10,16,26–28).
Combining cyclin-dependent kinase inhibitor 2A and retinoic acid
receptor β2 methylation yields sensitivity of 69% and specificity
of 87% (26). SHOX2 had a
sensitivity range of 68.0–75% and a specificity range of 94.0–95.0%
(10,27). Roncarati et al (28) reported that a four-gene methylation
panel [RASSF1A, cadherin 1, type 1, E-cadherin (epithelial),
DLC1 ρGTPase activating protein and peripherin] has high
sensitivity of 97.0% and a moderate specificity of 74.0%. Recently,
a prospective study was conducted using methylated homeobox A9 in
bronchial lavage among patients with suspected LC; results of the
study showed a sensitivity range of 73.1–80.0% and a specificity
range of 85.3–75.6%, with a PPV range of 90.7–78.4% and an NPV
range of 61.7–77.3% (16), which
are comparable with those in the present study. Another study
showed that the 23-gene expression classifier has the potential to
detect LC in bronchial epithelial cells collected during
bronchoscopy in two multicenter prospective studies with a high
sensitivity range of 88.0–89.0% and a low specificity of 47.0%
(29,30).
In the case of BW samples from patients with LC at
an early stage, a few malignant cells may be present among the
larger number of normal cells (10,28). A
highly sensitive and accurate detection method should be used to
measure neoplastic cell-specific biomarkers in BW specimens. In the
present study, LTE-qMSP test was optimized for the 3-plex system in
a closed single-tube to measure candidate methylation biomarkers.
The clinical performance of six methylation biomarker candidates
was evaluated using DNA from BW samples. Logistic regression
analysis was used to build a two-biomarker combination model,
comprising PCDHGA12 and PRRX1, as the best performing
biomarker for diagnosing LC. This two-biomarker combination model
test achieved a high sensitivity of 82.4% and a specificity of
87.9%, with a PPV of 93.3% and an NPV of 70.7%.
The present study has several limitations including
a small sample size that leads to insufficient statistical power,
lack of information on smoking history, an imbalance of the
male-to-female ratio of patients with LC (3.5:1.0), and the
retrospective case-control study design. Additionally, the
two-biomarker combination model test outperformed cytology in
sensitivity. However, combining the test with cytology did not
significantly improve diagnostic sensitivity. These data indicated
that the two-biomarker combination model has a high potential to
aid in diagnosing LC as an adjunct value to cytology using BW
samples.
The two-biomarker model exhibited lower sensitivity
for early-stage LC (I, II) than for late-stage LC (III, IV), which
may be attributed to a smaller number of the neoplastic cells in BW
samples (10). Notably, the results
of the present study indicate markedly higher sensitivity of the
two-biomarker model for patients with SCLC compared with that for
patients with NSCLC, comparable with research by Jeong et al
(31), which observed higher
sensitivity of PCDHGA12 for SCLC compared with that for
NSCLC. The reasons for this difference warrant further
investigation. In addition, the sensitivity for patients with
squamous cell carcinoma reached 90.0% but decreased to 66.7% for
patients with adenocarcinoma (ADC) in the present study. This
decline may be attributed to decreased shedding of ADC cells into
the airway (32).
The sensitivity of the two-biomarker model for
peripheral LC was significantly lower than that for LC in the
central region. At present, to overcome the limitations of standard
flexible bronchoscopy in its ability to detect small lung nodules
or peripheral lesions, radial-endobronchial ultrasound (R-EBUS) and
electromagnetic navigation bronchoscopy (ENB) have been introduced.
The American College of Chest Physicians guidelines for diagnosing
and managing LC recommend ENB or R-EBUS to evaluate peripheral lung
lesions that cannot be accessed with conventional flexible
bronchoscopy (33). Therefore, it
is worthwhile to investigate whether a two-biomarker test in BW
specimens in conjunction with R-EBUS or ENB can improve diagnostic
efficacy for patients with peripheral LC.
The present results underscore the potential use of
two aberrantly methylated genes, PCDHGA12 and PRRX1,
as effective biomarkers for non-invasive diagnostic tests aimed at
enhancing LC detection when used adjunctively with cytology in BW
specimens. In clinical practice, inconclusive bronchoscopy results
often require invasive procedures, potentially resulting in benign
diagnoses in a considerable number of cases, thus leading to cost
inefficiency. The proposed application of the methylation biomarker
test at an optimal cut-off value, coupled with cytology using BW
samples, demonstrates promising sensitivity. This test facilitates
identification of patients with a higher likelihood of harboring
malignancies, thereby guiding selection of candidates for invasive
bronchoscopy procedures. Moreover, the potential clinical benefits
of a two-biomarker test are evident in cases classified as
inconclusive, where a definitive cytological or histological
diagnosis of malignancy is lacking. If the test using two
methylation biomarkers is calibrated to optimal cut-off values,
resulting in high NPV, it could provide evidence that patients with
negative results may avoid unnecessary invasive procedures, thus
conferring benefits to patients in a cost-effective manner.
However, integrating the two-methylation biomarker model into
routine clinical practice requires rigorous validation through
large-scale prospective clinical trials.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank Mrs Yangyei Seo and
Mr. Jaejin Lee (Genomictree, Inc.), for their technical support
with DNA extraction from BW samples.
Funding
The present study was supported by Genomictree, Inc. (Daejeon,
South Korea; funding no. LC2-1-DEV).
Availability of data and materials
The data generated in the present study may be found
in the Gene Expression Omnibus under accession number GSE246510 or
at the following URL: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE246510.
Authors' contributions
SA conceived and designed the study. TJO, SHJ, SJK
and MAW acquired, analyzed and interpreted the data. IBJ, MHL and
JWS analyzed and interpreted data. TJO and SHJ confirm the
authenticity of all the raw data. TJO wrote the manuscript. SA
revised the manuscript for important intellectual content. SHJ, SJK
and MAW provided administrative, technical or material support. SA
supervised the study. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study adhered to local ethics guidelines
and was approved by the Institutional Review Board of Chungnam
National University Hospital (approval no. CNUH 2022-02-061-002)
and the Konyang University Hospital (approval no. 2022-03-025).
Written informed consent was obtained from all patients.
Patient consent for publication
Not applicable.
Competing interests
Genomictree, Inc., provided funding for the study.
TJO, SHJ, SJK, MAW and SA are employees of Genomictree, Inc. TJO
and SA are shareholders of Genomictree, Inc. The other authors
declare that they have no competing interests.
Glossary
Abbreviations
Abbreviations:
|
ADC
|
adenocarcinoma
|
|
AUC
|
area under the curve
|
|
BW
|
bronchial washing
|
|
LTE
|
linear target enrichment
|
|
MtI
|
methylation index
|
|
NPV
|
negative predictive value
|
|
NSCLC
|
non-small cell lung cancer
|
|
PCDHGA12
|
protocadherin γ, subfamily A, 12
|
|
PPV
|
positive predictive value
|
|
PRRX1
|
paired-related homeobox 1
|
|
qMSP
|
quantitative methylation-specific
PCR
|
|
ROC
|
receiver operating characteristic
|
|
SCC
|
squamous cell carcinoma
|
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soeromataram I, Jemal A and Bray F: Global caner statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tomasik B, Skrzypski M, Bieńkowski M,
Dziadziuszko R and Jassem J: Current and future applications of
liquid biopsy in non-small-cell lung cancer-a narrative review.
Trans Lung Cancer Res. 12:594–614. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Blandin Knight S, Crosbie PA, Balata H,
Chudziak J, Hussell T and Dive C: Progress and prospects of early
detection in lung cancer. Open Biol. 7:1700702017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tsou JA, Galler JS, Siegmund KD, Laird PW,
Turla S, Cozen W, Hagen JA, Koss MN and Laird-Offringa IA:
Identification of a panel of sensitive and specific DNA methylation
markers for lung adenocarcinoma. Mol Cancer. 6:702007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Oken MM, Hocking WG, Kvale PA, Andriole
GL, Buys SS, Church TR, Crawford ED, Fouad MN, Isaacs C, Reding DJ,
et al: Screening by chest radiograph and lung cancer mortality: The
prostate, lung, colorectal, and ovarian (PLCO) randomized trial.
JAMA. 306:1865–1873. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
The National Lung Screening Trial Research
Team, . Aberle DR, Adams AM, Berg CD, Black WC, Clapp JD,
Fagerstrom RM, Gareen IF, Gatsonis C, Marcus PM and Sicks JD:
Reduced lung-cancer mortality with low-dose computed tomographic
screening. New Eng J Med. 365:395–409. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang C, Yu W, Wang L, Zhao M, Guo Q, Lv
S, Hu X and Lou J: DNA methylation analysis of the SHOX2 and
RASFF1A panel in bronchoavelolar lavage fluid for lung cancer
diagnosis. J Cancer. 8:3585–3591. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hu S, Tao J, Peng M, Ye Z, Chen Z, Chen H,
Yu H, Wang B, Fan J and Ni B: Accurate detection of early-stage
lung cancer using a panel of circulating cell-free DNA methylation
biomarkers. Biomarker Res. 11:452023. View Article : Google Scholar
|
|
9
|
Tsou JA, Hagen JA, Carpenter CL and
Laird-Offringa IA: DNA methylation analysis: A powerful new tool
for lung cancer diagnosis. Oncogene. 21:5450–5461. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schmidt B, Liebenberg V, Dietrich D,
Schlegel T, Kneip C, Seegebarth A, Flemming N, Seemann S, Distler
J, Lewin J, et al: SHOX2 DNA methylation is a biomarker for
the diagnosis of lung cancer based on bronchial aspirates. BMC
Cancer. 10:6002010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dietrich D, Kneip C, Raji O, Liloglou T,
Seegebarth A, Schlegel T, Flemming N, Rausch S, Distler J,
Fleischhacker M, et al: Performance evaluation of the DNA
methylation biomarker SHOX2 for the aid in diagnosis of lung
cancer based on the analysis of bronchial aspirates. Int J Oncol.
40:825–832. 2012.PubMed/NCBI
|
|
12
|
Wiener RS, Wiener DC and Gould MK: Risks
of transthoracic needle biopsy: How high? Clin Plum Med. 20:29–35.
2013. View Article : Google Scholar
|
|
13
|
Li P, Liu S, Du L, Mohseni G, Zhang Y and
Wang C: Liquid biopsies based on DNA methylation as biomarkers for
the detection and prognosis of lung cancer. Clin Epigenetics.
14:1182022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ma S, Yu X, Jin X, Qiu F, Chen X, Wang R
and Cao C: The usefulness of liquid-based cytology of
bronchoalveolar lavage fluids combined with bronchial brush
specimens in lung cancer diagnosis. J Int Med Res.
50:30006052211327082022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chrabańska M, Środa M, Kiczmer P and
Droddzowska B: Lung cancer cytology: Can any of the cytological
methods replace histopathology? J Cytol. 37:117–1121. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wen SWC, Wen J, Hansen TF and Jakobsen A:
Cell free methylated tumor DNA in bronchial lavage as an additional
tool for diagnosing lung cancer-a systematic review. Cancers
(Basel). 14:22542022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liang V, Li X, Li W, Zhu X and Li C: DNA
methylation in lung cancer patients: Opening a ‘window of life’
under precision medicine. Biomed Pharmacother. 144:1122022021.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Esteller M: Epigenetics in cancer. N Eng J
Med. 358:1148–1159. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ibrahim J, Peeters M, Van Camp G and Op de
Beeck K: Methylation biomarkers for early cancer detection and
diagnosis: Current and future perspectives. Eur J Cancer.
178:91–113. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Anglim PP, Alonzo TA and Laird-Offringa
IA: DNA methylation-based biomarkers for early detection of
non-small cell lung cancer: An update. Mol Cancer. 7:812008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Z, Xie K, Zhu G, Ma C, Cheng C, Li Y,
Xiao X, Li C, Tang J, Wang H, et al: Early detection and
stratification of lung cancer aided by a cost-effective assay
targeting circulating tumor DNA (ctDNA) methylation. Respir Res.
24:1632023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rauch T, Wang Z, Zhang X, Zhong X, Wu X,
Lau SK, Kernstine KH, Riggs AD and Pfeifer GP: Homeobox gene
methylation in lung cancer studied by genome-wide analysis with a
microarray-based methylated CpG island recovery assay. Proc Natl
Acad Sci USA. 104:5527–5532. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hong Y and Kim WJ: DNA methylation markers
in lung cancer. Curr Genomics. 22:79–87. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dejeux E, Audard V, Cavard C, Gut IG,
Terris B and Tost J: Rapid identification of promoter
hypermethylation in hepatocellular carcinoma by pyrosequencing of
etiologically homogeneous sample pools. J Mol Diag. 9:510–520.
2007. View Article : Google Scholar
|
|
25
|
Chung W, Bondaruk J, Jelinek J, Lotan Y,
Liang S, Czerniak B and Issa JJ: Detection of bladder cancer using
novel DNA methylation biomarkers in urine sediments. Cancer
Epidemiol Biomarkers Prev. 20:1483–1491. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Grote HJ, Schiemann V, Gedder H, Rohr UP,
Kappes R, Gabbert HE and Böcking A: Aberrant promoter methylation
of p16(INKA4a), RARB2 and SEMA3B in bronchial aspirates from
patients with suspected lung cancer. Int J Cancer. 116:720–725.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ni S, Ye M and Huang T: Short stature
homeobox 2 methylation as a potential noninvasive biomarker in
bronchial aspirates for lung cancer diagnosis. Oncotarget.
8:61253–61263. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Roncarati R, Lupini L, Miotto E, Saccenti
E, Mascetti S, Morandi L, Bassi C, Rasio D, Callegari E, Conti V,
et al: Molecular testing on bronchial washings for the diagnosis
and predictive assessment of lung cancer. Mol Oncol. 14:2163–2175.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Silvestri GA, Vachani A, Whitney D,
Elashoff M, Porta Smith K, Ferguson JS, Parsons E, Mitra N, Brody
J, Lenburg ME, et al: A bronchial genomic classifier for the
diagnostic evaluation of lung cancer. N Eng J Med. 373:243–251.
2015. View Article : Google Scholar
|
|
30
|
Vachani A, Whitney DH, Parsons EC, Lenburg
M, Ferguson JS, Silvestri GA and Spira A: Clinical utility of a
bronchial genomic classifier in patients with suspected lung
cancer. Chest. 150:210–218. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jeong IB, Yoon YS, Park SY, Cha EJ, Na MJ,
Kwon SJ, Kim JH, Oh TJ, An S, Park CR, et al: PCDHGA12
methylation biomarkers in bronchial washing specimens as an
adjunctive diagnostic tool to bronchoscopy in lung cancer. Oncol
Lett. 16:1039–1045. 2018.PubMed/NCBI
|
|
32
|
Ahrendt SA, Chow JT, Xu L, Yang SC,
Eisenberger CF, Esteller M, Herman JG, Wu L, Decker PA, Jen J and
Sidransky D: Molecular detection of tumor cells in bronchoalveolar
lavage fluid from patients with early stage lung cancer. J Natl
Cancer Inst. 91:332–339. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liam CK, Lee P, Yu CJ, Bai C and Yasufuku
K: The diagnosis of lung cancer in the era of interventional
pulmonology. Int J Tuberc Lung Dis. 25:6–15. 2021. View Article : Google Scholar : PubMed/NCBI
|














