Introduction
Atherosclerosis is an inflammatory disease driven by
the interaction of inflammatory cells and the vascular wall
(1,2). Vascular neointimal hyperplasia is a
key event in arteriosclerosis and several other vascular diseases
(3,4). Neointimal hyperplasia is an abnormal
increase in the cell population within the innermost layer of an
arterial wall (5). The underlying
causes of neointimal hyperplasia include the migration,
proliferation and apoptosis of vascular smooth muscle cells (VSMCs)
provoked by endovascular injury or perivascular injury (6,7).
According to the mechanisms above, the neointima consists mainly of
VSMCs and the migration and proliferation of VSMCs are critical to
neointimal hyperplasia following arterial injury (8). Complement components have been
identified in neointimal plaques and atherosclerotic tissues
(9,10). Complement anaphylatoxin C5a is a
risk factor for atherosclerosis progression. Following arterial
injury, C5a leads to pro-inflammatory activation and neointima
formation during arterial remodeling (11). C5a affects the migration and
proliferation of VSMCs through different pathways. C5a is able to
bind to the C5a receptor on VSMCs and induce the autocrine
signaling of factors, including vascular cell adhesion molecule-1
(VCAM-1) and platelet derived growth factor (PDGF) (11). It also binds to the receptor on
macrophages and provokes the production of large amounts of
cytokines and chemokines, including tumor necrosis factor-α
(TNF-α), interleukin-6 (IL-6) and monocyte chemotactic protein-1
(MCP-1) (12), that accelerate
neointimal hyperplasia by promoting the migration and proliferation
of VSMCs (13).
C4a and C5a are liberated from the N-terminal region
of the parental protein α-chain. Although they are similar in terms
of molecular structure, it has been demonstrated that C5a is able
to act on numerous types of cells; however, C4a is restricted to
monocytes/macrophages in phagocytic leukocytes (14). The interactions between C5a and
other complement anaphylatoxins have been investigated in numerous
fields; however, the interactive role of C4a and C5a during
arterial remodeling following injury has not been studied. Since
C5a is able to promote the migration and proliferation of VSMCs via
a macrophage-mediated inflammatory reaction, and C4a is able to
inhibit the chemoattractant and secretagogue induced by C5a in
monocytes/macrophages (15), the
present study sought to determine if C4a inhibits C5a-induced
neointima formation following arterial injury in the complement
cascades.
In the present study, to the best of our knowledge,
the hypothesis that C4a inhibits C5a-induced neointima formation
following arterial injury was tested for the first time, and the
results indicated that this may be a novel therapeutic strategy for
neointimal hyperplasia.
Materials and methods
Animals
Animal care and protocol for this study were in
accordance with the Animal Experiment Guidelines of Harbin Medical
University (Harbin, China) and ethical approval was obtained from
Harbin Medical University. C57BL/6N, 16-week-old mice (Fuzhong
Biotechnology, Shanghai, China) were subjected to transluminal
endovascular wire injury of the bilateral femoral artery, as
reported previously (8). All the
mice were fed normal chow and water ad libitum and kept
under normal diurnal cycle at room temperature (22°C). Over a
course of two weeks, recombinant C5a (Sigma, Shanghai, China; 1
μg/25 g of body weight) and recombinant C4a (1 μg/25 g of body
weight) were administered via an Alzet mini-osmotic pump (American
Health & Medical Supply International Corp, Chengdu, Sichuan,
China) into the subcutaneous perifemoral artery following
endovascular injury, according to a previously described method
(16). Image analysis software was
used to measure the neointima and media of the femoral artery. The
relative mRNA expression levels of CD68 and F4/80 in femoral
arterial tissue were assessed by quantitative polymerase chain
reaction (qPCR), and the protein levels of TNF-α, IL-6 and MCP-1 in
femoral arterial tissue were assessed by western blot analysis.
Preparation of recombinant C4a
C4a cDNA was prepared by PCR using total mRNA
derived from HepG2 cells. The confirmed nucleotide sequence of the
PCR product was subcloned into the expression vector pET32a flanked
by BamHI and EcoRI restriction sites, and each
plasmid cDNA was transformed to DH5α competent cells. Rosetta-gami
B (DE3) Lys-S cells transformed with each expression vector were
cultured in Lennox Broth medium containing ampicillin,
chloramphenicol, kanamycin and tetracycline until the 600 nm
absorbance of the bacterial suspension reached 0.6. Each suspension
was mixed with 1 mM of isopropyl 1-thio-β-d-galactoside and
incubated for an additional 4 h. Following centrifugation, the
cultured E. coli cells were resuspended into 1/10 culture volume
(50 ml) of 20 mM Tris-HCl containing 200 mM NaCl and 10 mM EDTA (pH
8.0). The bacteria were lysed by sonication in the presence of 1%
Triton X-100. Following centrifugation, the extracted recombinant
proteins were separated using a Hi-Trap™ Chelating HP column
preloaded with 100 mM NiSO4 (GE Healthcare Life
Sciences, Piscataway, NJ, USA). The purity of Trx-His-S-tag
recombinant C4a was checked using SDS-PAGE. The recombinant C4a was
dialyzed in phosphate-buffered saline (PBS) containing 1 mg/ml of
bovine serum albumin (BSA; Sigma) and stored at −70°C until
use.
Cells
Primary culture of VSMCs
After the C57BL/6N mice were sacrificed, the aortas
were separated and transferred into 10 cm dishes containing
Dulbecco’s modified Eagle’s medium (DMEM). The endothelium was
injured by prodding three to five times with a sterilized cotton
swab. Endothelium-injured aortas were placed down and then cut into
0.2×0.2 cm squares, which were carefully transferred into six-well
multiplates, two to three squares per well, containing DMEM + 20%
fetal calf serum (FCS) culture medium (Sigma). Three to five
generations of VSMCs were used in this experiment. VSMCs were
exposed to C5a (10−8 M) in the presence or absence of
C4a (10−8 M) or exposed to the conditioned medium of
macrophages that had been pretreated with C5a in the presence or
absence of C4a, following which the migration and proliferation of
VSMCs were investigated and VCAM-1 expression was assessed by flow
cytometry.
Primary culture of peritoneal
macrophages
C57BL/6N mice were injected intraperitoneally with
4.0 ml of 4% thioglycollate (Difco Laboratories, Franklin Lakes,
NJ, USA) and peritoneal macrophages were isolated from peritoneal
lavage liquid three days later. Peritoneal macrophages were
cultured in RPMI-1640 medium supplemented with 10% FCS for three
days. Subsequently, C5a (10−8 M) was added to untreated
macrophages or those that had been pretreated with C4a
(10−8 M) for 10 min. Then, TNF-α, IL-6 and MCP-1
expression, Ca2+ influx and ERK activation in
macrophages were assayed, respectively, by ELISA, a calcium imaging
system and western blot analysis.
Quantification of neointimal
hyperplasia
The mice were sacrificed and the femoral arteries
were harvested two weeks after surgical intervention. Arterial
tissue was fixed in 10% formalin and embedded in paraffin. The
middle segment of the artery was cut into five serial
cross-sections at 200 μm intervals. Following elastica van Gieson
staining for connective tissue, areas of the neointima and artery
were measured using image analysis software (Image J; http://imagej.nih.gov/ij/), as previously described
(17).
qPCR
Total RNA was extracted from femoral arterial tissue
and relative mRNA was normalized to 18s. The following primers were
used: F4/80 forward, 5′-GAGATTGTGGAAGCATCCGAGAC-3′ and reverse,
5′-GATGACTGTACCCACATGGCTGA-3′; Cd68 forward,
5′-CATCAGAGCCCGAGTACAGTCTACC-3′ and reverse,
5′-AATTCTGCGCCATGAATGTCC-3′; 18s forward,
5′-GTAACCCGTTGAACCCCATT-3′ and reverse, 5′-CCATCCAATCGGTAGTAGCG-3′.
qPCR was performed using the ABI 7300 Fast real-time PCR system
(Applied Biosystems, Foster City, CA, USA).
Western blot analysis
Electrophoresis was performed using a vertical slab
gel with a 12% polyacrylamide content according to the method by
Laemmli (18). The transfer of
proteins from the SDS polyacrylamide gel to a membrane was
performed electrophoretically according to the method by
Kyhse-Andersen (19) with certain
modifications using a Semi Dry Electroblotter (Sartorius AG,
Goettingen, Germany) for 90 min with an electric current of 15 V.
The membrane was treated with Block Ace™ (4%) for 30 min at 22°C.
The first reaction was performed using rabbit immunoglobulin (IG) G
antibodies against TNF-α, IL-6, MCP-1 and unmodified protein or
against phosphorylated protein of ERK 1/2 (100 ng/ml; Sigma) in PBS
containing 0.03% Tween-20 for 1 h at 22°C. Following washing in the
same buffer, the second reaction was performed using horseradish
peroxidase (HRP)-conjugated anti-rabbit goat IgG (20 ng/ml) for 30
min at 22°C. Following washing, the enhanced chemiluminescence
(ECL) reaction was performed on the membrane using the ECL Plus
Western Blotting Detection System™ (GE Healthcare Life
Sciences).
Flow cytometry
VSMCs (2×106 cells/ml) were incubated for
24 h with C5a (10−8 M) in the presence or absence of C4a
(10−8 M) or with conditioned medium of macrophages that
had been pretreated with C5a in the presence or absence of C4a.
VSMCs were stained with anti-mouse CD106 (VCAM-1)-phycoerythrin rat
IgG2a (Sigma) antibodies as the isotype control for 30 min on ice.
The cells were analyzed using a FACS Calibur flow cytometer (BD
Biosciences, Tokyo, Japan).
Migration assay
The migration assay was performed using a 48-well
chamber migration assay kit with Nuclepore filters (Nuclepore,
Pleasanton, CA, USA) with a pore size of 8 μm according to the
method by Falk et al (20).
For preparation, the upper wells were coated with 0.01% collagen
for 30 min of incubation at 37°C. Next, VSMCs (5×104
cells/well) were seeded into the top wells. The chemotactic medium
was added to the lower wells. Following being incubated at 37°C for
8 h, the cells that had migrated to a lower filter surface were
fixed with 4% paraformaldehyde in PBS for 10 min at room
temperature and stained with hematoxylin and eosin Y. Cell
migration was defined as the number of cells that had migrated to a
lower filter surface.
Proliferation assay
The proliferation assay was performed by using the
Cell Counting kit-8 (CCK-8; Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan) according to the manufacturer’s instructions.
Firstly, a suspension of VSMCs (5,000 cells/100 μl/well) was loaded
into the wells of a 96-well plate. The cells were then incubated at
37°C for 24 h. CCK-8 solution (10 μl) was added to each well and
the cells were incubated for 3 h at 37°C. A microplate reader was
then used to measure the absorbance at 450 nm. According to the
prepared standard curve, the relative cell numbers were
calculated.
ELISA
C5a (10−8 M) was added to untreated
macrophages or those that had been pretreated with C4a
(10−8 M) for 10 min. Then, the supernatant was collected
for analysis of TNF-α, IL-6 and MCP-1 by using the mouse ELISA kit
(Sigma). The ELISA plates were coated with 100 μl capture antibody
per well at 4°C overnight. Following an appropriate wash, 200 μl of
assay dilution buffer was added per well for inhibition at room
temperature for 1 h. The sample and serial dilutions of the
standards were added to the plate and incubated at 4°C overnight.
Following coating with detection antibody, avidin-HRP was added and
incubated at room temperature for 30 min. The substrate
3,3′,5,5′-tetramethylbenzidine was added and incubated for 15 min.
Finally, 2N H2SO4 was added to terminate the
reaction and the absorbance at 450 nm was measured using an ELISA
reader (MTP-800 Microplate reader; Corona Electric, Tokyo,
Japan).
Measurement of cytoplasmic
Ca2+ influx
Ca2+ imaging was performed as described
previously (21). The macrophages
(2×106 cells/ml) were loaded with calcium-sensitive Fura
2-AM (1 μM) in Ca2+-free buffer (Hanks’ balanced salt
solution containing 20 mM of
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid and 1% BSA; pH
7.4) for 30 min at 37°C according to the manufacturer’s
instructions (Dojindo Laboratories, Inc.). The samples were placed
directly into the cell suspension following a 3 min baseline
recording. The recordings were made with an F-2500 calcium imaging
system from FL Solutions (Hitachi, Tokyo, Japan) that calculated
the ratio of fluorescent signals obtained at 37°C with excitation
wavelengths at 340 and 380 nm, and with an emission wavelength at
510 nm. The excitation wavelengths at 380 nm and 340 nm were used
to measure the free Fura-2 and the Ca2+-bound Fura-2,
respectively. The fluorescent activities of 340 nm/500 nm (F1) and
of 380 nm/500 nm (F2) and the ratio (R) of F1 to F2 were recorded
by the spectrophotometer at the indicated time. The Ca2+
concentration (C) was then calculated using the following formula:
C = 224 × R, where 224 is the Kd number.
Statistical analysis
Data are expressed as the mean ± standard error or
mean ± standard deviation. Each experiment was repeated at least
three times. The Student’s t-test was used and P<0.05 was
considered to indicate a statistically significant difference.
Results
Inhibitory effect of C4a on C5a-induced
neointima formation
To investigate the inhibitory effect of C4a on
C5a-induced neointima formation, the mice were treated with C5a (1
μg/25 g of body weight) or C5a + C4a (1 μg/25 g of body weight) by
mini-osmotic pumps for two weeks after injury. Following treatment
with C5a, the neointimal area increased significantly (Fig. 1; P<0.01). C5a also increased the
mRNA expression of CD68 and F4/80 (Fig. 2A and B; P<0.01) and the protein
levels of TNF-α, IL-6 and MCP-1 (Fig.
2C; P<0.01) in femoral artery tissue. However, following C5a
and C4a treatment, the neointimal area was significantly reduced
(Fig. 1; P<0.05). C4a also
inhibited the mRNA expression of CD68 and F4/80 (Fig. 2A and B; P<0.05) and the protein
levels of TNF-α, IL-6 and MCP-1 (Fig.
c; P<0.05) in femoral artery tissue induced by C5a
stimulation.
 | Figure 2Inhibitory effect of C4a on
C5a-induced mRNA expression of CD68 and F4/80 and the protein
levels of TNF-α, IL-6 and MCP-1 in femoral artery tissue. Mice were
treated with C5a (1 μg/25 g of body weight) or C5a + C4a (1 μg/25 g
body weight) by mini-osmotic pumps for two weeks after injury.
Following treatment with C5a, the mRNA expression of (A) CD68, (B)
F4/80 as well as the protein levels of (C) TNF-α, IL-6 and MCP-1
increased significantly. The increased mRNA expression of (A) CD68,
(B) F4/80 and protein levels of (C) TNF-α, IL-6 and MCP-1 were
significantly reduced following C4a treatment. Data are expressed
as the mean ± standard error (n=5). P<0.05 was considered to
indicate a statistically significant difference.
(**P<0.01, *P<0.05, injury + C5a vs
injury; ##P<0.01, #P<0.05, injury +
C5a/C4a vs injury + C5a). TNF-α, tumor necrosis factor-α;
interleukin-6; MCP-1, monocyte chemotactic protein-1. |
Lack of inhibitory effect of C4a on VSMCs
in response to C5a
Since VSMCs are critical in vascular remodeling
following vascular injury (22),
the effect of C4a on the VCAM-1 regulation of VSMCs was
investigated. VSMCs were treated with C5a (10−8 M) in
the presence or absence of C4a (10−8 M), then the
migration, proliferation and VCAM-1 expression of VSMCs were
investigated. C4a did not inhibit C5a-induced low levels of the
migration (Fig. 3A), proliferation
(Fig. 3B) or VCAM-1 expression of
VSMCs (Fig. 3C).
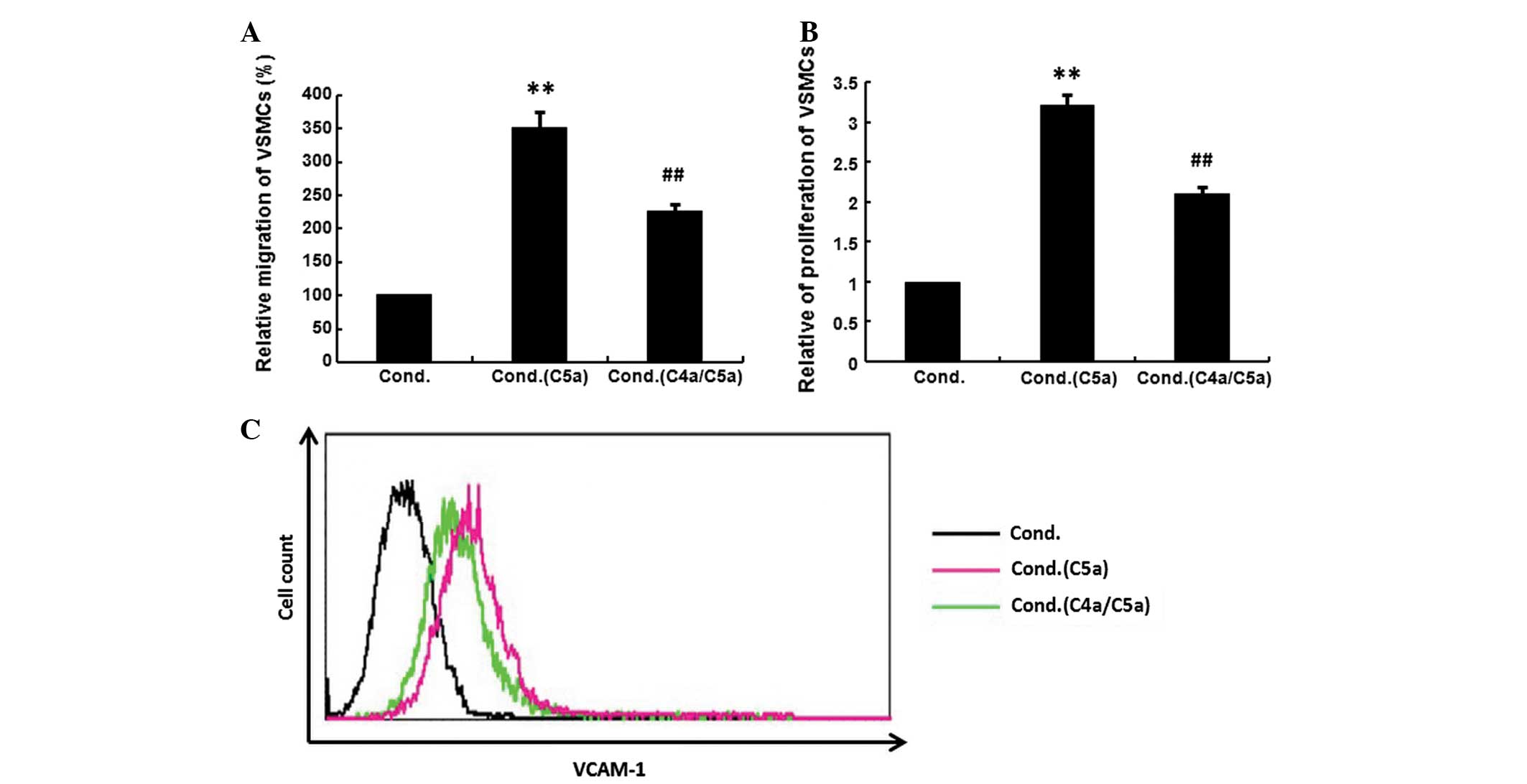
Inhibition of VSMCs by conditioned medium
from C4a-pretreated macrophages
C5a (10−8 M) was added to untreated
macrophages or those that had been pretreated with C4a
(10−8 M), then the supernatant from macrophages was
added to VSMCs. The migration, proliferation and VCAM-1 expression
of VSMCs were investigated. The conditioned medium from C5a-treated
macrophages significantly increased the migration, proliferation
and VCAM-1 expression of VSMCs. The increased migration,
proliferation and the upregulation of VCAM-1 expression were
significantly suppressed when VSMCs were exposed to the conditioned
medium from C4a-pretreated macrophages (Fig. 4; P<0.01).
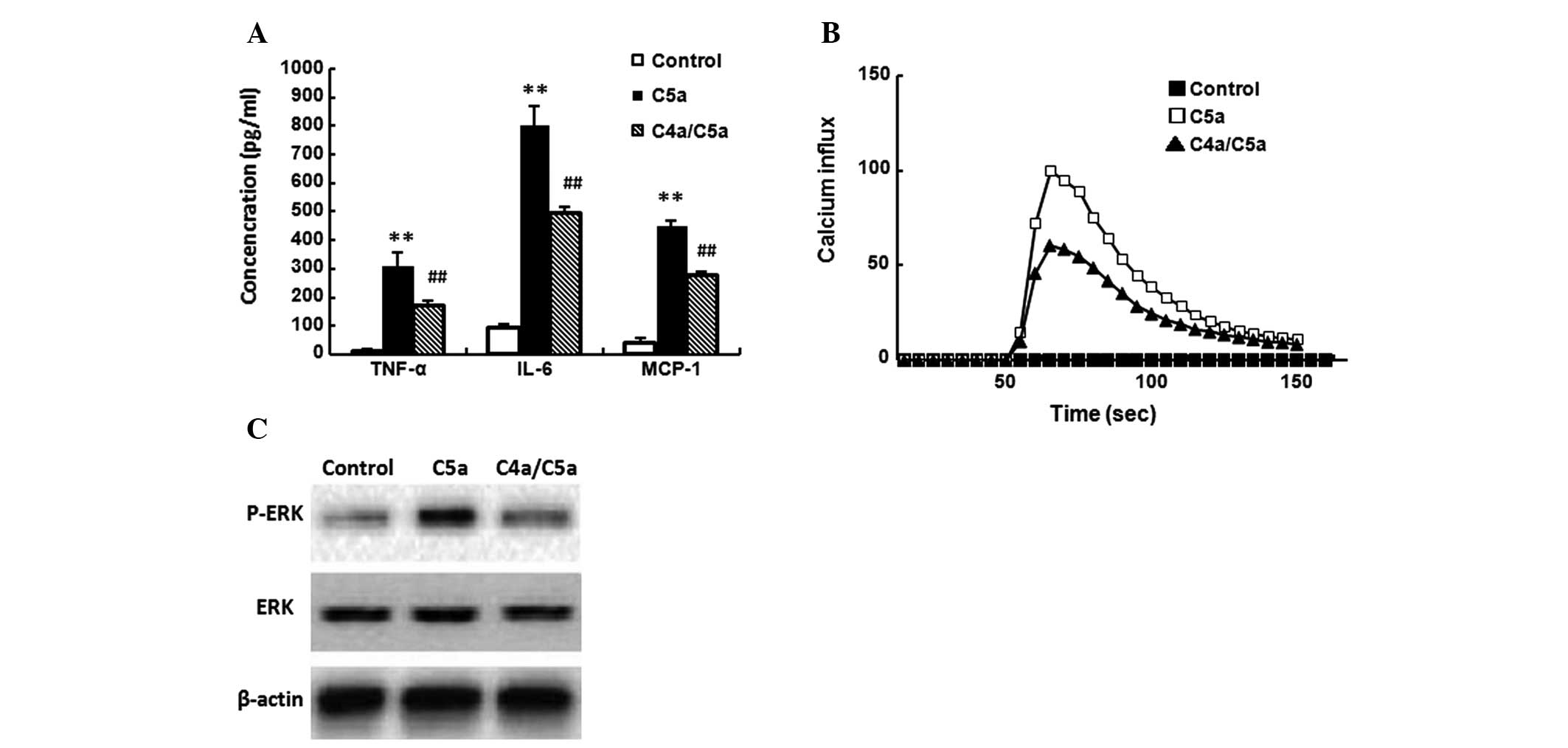
Inhibitory effect of C4a on macrophages
in response to C5a
C5a (10−8 M) was added to untreated
macrophages or those that had been pretreated with C4a
(10−8 M) for 10 min. Then, TNF-α, IL-6 and MCP-1
expression, Ca2+ influx and ERK activation in
macrophages were assayed, respectively. C5a significantly induced
TNF-α, IL-6 and MCP-1 expression (Fig.
5A), Ca2+ influx (Fig.
5B) and ERK activation (Fig.
5C) in macrophages (P<0.01). C4a significantly suppressed
C5a-induced TNF-α, IL-6 and MCP-1 expression and Ca2+
influx (Fig. 5A and B; P<0.01),
and inhibited C5a-induced ERK activation (Fig. 5C). C4a alone did not induce the
expression of TNF-α, IL-6 and MCP-1, Ca2+ influx or
phosphorylation of ERK (data not shown).
Discussion
The present study demonstrated, for the first time,
to the best of our knowledge, that the inhibition of C5a by C4a
limits neointima formation following arterial injury in
wire-induced endothelial denudation of the femoral artery of mice.
Although C4a and C5a are released from the N-terminal region of the
parental protein α-chain and are similar in terms of molecular
structure, they have different functions (23). C5a is a potent soluble
anaphylotoxic and chemotactic inflammatory mediator (24). It is able to act on numerous types
of cells by binding to the C5a receptor (25); as such, C5a induces
pro-inflammatory activation and targets neointima formation
following arterial injury (11).
C4a was previously isolated from the inflammatory joint fluid of
patients with rheumatoid arthritis and identified as the
monocyte/macrophage migration inhibitory factor (26). The C4a receptor is restricted to
monocytes/macrophages in phagocytic leukocytes (14). C4a is able to inhibit the
chemoattractant and secretagogue induced by C5a in
monocytes/macrophages (15). The
present study investigated whether C4a was able to inhibit
C5a-induced neointima formation via the complement cascade. The
present study confirmed the stimulatory effect of C5a on neointima
formation and demonstrated the protective effect of C4a treatment
following injury.
VSMCs from the media are key in the progressive
intimal thickening that leads to atherosclerosis and restenosis
(22). Since VSMCs are critical in
neointimal hyperplasia, the present study focused on those factors
that are able to affect VSMCs. C5a is a risk factor for the
progression of atherosclerosis (27) It affects the migration and
proliferation of VSMCs through different pathways by a series of
coordinated signals (11,28,29).
C5a is able to bind to the C5a receptor on VSMCs (29) and induce the autocrine signaling of
factors, including VCAM-1 and PDGF (11). VCAM-1 and PDGF are important in
early atherogenesis, their expression is a marker of the transition
of VSMCs to the synthetic phenotype within atherosclerotic lesions
(30). Following arterial injury,
inflammatory cells, including monocytes and macrophages, are
recruited into neointimal sites (31). Consistent with that, our data
demonstrated that the mRNA expression of CD68 and F4/80 (as
indicators of macrophage accumulation) in femoral artery tissue
increases significantly following treatment with C5a (Fig. 2A and B). C5a also binds to the
receptors on inflammatory cells, particularly on macrophages, and
provokes the production of large amounts of cytokines and
chemokines, including TNF-α, IL-6 and MCP-1 (Fig. 2C) (12). TNF-α is a canonical inflammatory
cytokine that promotes the migration and proliferation of VSMCs
(13). IL-6 is a pleiotropic
cytokine involved in pro-inflammatory and anti-inflammatory
responses via regulating leukocyte function and apoptosis (32). MCP-1 is a potent chemoattractant
that is essential in various inflammatory diseases involving the
recruitment of monocytes/macrophages (33). These factors all accelerate
neointimal hyperplasia by promoting the migration and proliferation
of VSMCs (13,34).
With the receptor on VSMCs, C5a induced low levels
of migration and proliferation as well as VCAM-1 expression in
VSMCs; however, this migration, proliferation and VCAM-1 expression
could not be suppressed by C4a. These results demonstrated that C4a
did not directly act on VSMCs. There is no evidence verifying the
presence of a VSMC C4a receptor and our data also suggest its
absence in VSMCs. However, when C5a was added to untreated or
C4a-pretreated macrophages and then the supernatant from
macrophages was added to VSMCs, it was revealed that C5a-treated
macrophages-conditioned medium, which contained cytokines TNF-α,
IL-6 and chemokine MCP-1, significantly increased the migration and
proliferation of VSMCs, and TNF-α further increased VCAM-1
expression in VSMCs. These results were consistent with previous
studies (12,35). The increased migration,
proliferation and upregulation of VCAM-1 expression were
significantly suppressed when VSMCs were exposed to the conditioned
medium from C4a-pretreated macrophages (Fig. 4). Regarding the underlying
mechanism, the present study revealed that the C5a-induced
expression of TNF-α, IL-6 and MCP-1 in macrophages was suppressed
by C4a (Fig. 5A). These results
confirmed the presence of a C4a receptor on macrophages.
It has been demonstrated that the release of
cytokines and chemokines from C5a-stimulated macrophages is
associated with cytoplasmic Ca2+ influx (36). C5a binds to the C5a-receptor and
induces cytoplasmic Ca2+ influx (37), thereby stimulating the release of
TNF-α, IL-6 and MCP-1 from macrophages (38). In order to better understand the
protective mechanisms of C4a on C5a-induced neointima formation,
the present study examined whether C4a was able to inhibit
C5a-induced cytoplasmic Ca2+ influx in macrophages. C4a
significantly suppressed cytoplasmic Ca2+ influx in
macrophages (Fig. 5B).
Furthermore, as the ERK1/2 pathway is a process that includes the
authentic cytoplasmic Ca2+ influx (39), whether C4a was able to inhibit
C5a-induced ERK activation in macrophages was investigated. It was
observed that C4a effectively inhibited the ERK1/2 pathway, which
included Ca2+ mobilization (Fig. 5C). These in vitro data are
in accordance the in vivo findings of the present study.
According to the mechanism described in Fig. 6, C4a inhibits C5a-induced neointima
formation, not by acting directly on VSMCs, but via a
macrophage-mediated inflammatory reaction by inhibiting the
Ca2+-dependent ERK signaling pathway in macrophages. The
complement cascade is a complex process, although the inhibition of
C5a-induced neointima formation by C4a was demonstrated, the
complex inhibitory process and the identification of a C4a-receptor
requires further study.
The present study, to the best of our knowledge, is
the first demonstration that treatment with C4a significantly
reduces C5a-induced neointima formation following arterial injury,
via a macrophage-mediated inflammatory reaction by inhibiting the
Ca2+-dependent ERK signaling pathway in macrophages. The
crucial role of C5a/C4a reactions in neointima formation following
vascular injury is expected to provide important information for
the development of novel clinical treatments for vascular
diseases.
References
|
1
|
Ivanov V, Cha J, Ivanova S, Kalinovsky T,
Rath M and Niedzwiecki A: Nutrient supplementation modulates
angiotensin II-mediated atherosclerosis in ApoE KO mice. Mol Med
Rep. 3:417–425. 2010.PubMed/NCBI
|
|
2
|
Packard RR, Lichtman AH and Libby P:
Innate and adaptive immunity in atherosclerosis. Semin
Immunopathol. 31:5–22. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mayr M and Xu Q: Smooth muscle cell
apoptosis in arteriosclerosis. Exp Gerontol. 36:969–987. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dzau VJ, Braun-Dullaeus RC and Sedding DG:
Vascular proliferation and atherosclerosis: new perspectives and
therapeutic strategies. Nat Med. 8:1249–1256. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Newby AC and Zaltsman AB: Molecular
mechanisms in intimal hyperplasia. J Pathol. 190:300–309. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stone JD, Narine A, Shaver PR, Fox JC,
Vuncannon JR and Tulis DA: AMP-activated protein kinase inhibits
vascular smooth muscle cell proliferation and migration and
vascular remodeling following injury. Am J Physiol Heart Circ
Physiol. 304:H369–H381. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Korshunov VA and Berk BC: Smooth muscle
apoptosis and vascular remodeling. Curr Opin Hematol. 15:250–254.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sata M, Maejima Y, Adachi F, Fukino K,
Saiura A, Sugiura S, Aoyagi T, Imai Y, Kurihara H, Kimura K, Omata
M, Makuuchi M, Hirata Y and Nagai R: A mouse model of vascular
injury that induces rapid onset of medial cell apoptosis followed
by reproducible neointimal hyperplasia. J Mol Cell Cardiol.
32:2097–2104. 2000. View Article : Google Scholar
|
|
9
|
Niculescu F and Rus H: The role of
complement activation in atherosclerosis. Immunol Res. 30:73–80.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shagdarsuren E, Bidzhekov K, Djalali-Talab
Y, Liehn EA, Hristov M, Matthijsen RA, Buurman WA, Zernecke A and
Weber C: C1-esterase inhibitor protects against neointima formation
after arterial injury in atherosclerosis-prone mice. Circulation.
117:70–78. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shagdarsuren E, Bidzhekov K, Mause SF,
Simsekyilmaz S, Polakowski T, Hawlisch H, Gessner JE, Zernecke A
and Weber C: C5a receptor targeting in neointima formation after
arterial injury in atherosclerosis-prone mice. Circulation.
122:1026–1036. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pushparaj PN, Tay HK, Wang CC, Hong W and
Melendez AJ: VAMP8 is essential in anaphylatoxin-induced
degranulation, TNF-alpha secretion, peritonitis, and systemic
inflammation. J Immunol. 183:1413–1418. 2009. View Article : Google Scholar
|
|
13
|
Rectenwald JE, Moldawer LL, Huber TS,
Seeger JM and Ozaki CK: Direct evidence for cytokine involvement in
neointimal hyperplasia. Circulation. 102:1697–1702. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tsuruta T, Yamamoto T, Matsubara S,
Nagasawa S, Tanase S, Tanaka J, Takagi K and Kambara T: Novel
function of C4a anaphylatoxin. Release from monocytes of protein
which inhibits monocyte chemotaxis. Am J Pathol. 142:1848–1857.
1993.PubMed/NCBI
|
|
15
|
Murakami Y, Yamamoto T, Imamichi T and
Nagasawa S: Cellular responses of guinea pig macrophages to C4a;
inhibition of C3a-induced O2-generation by C4a. Immunol Lett.
36:301–304. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Takaoka M, Nagata D, Kihara S, Shimomura
I, Kimura Y, Tabata Y, Saito Y, Nagai R and Sata M: Periadventitial
adipose tissue plays a critical role in vascular remodeling. Circ
Res. 105:906–911. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Miyata K, Oike Y, Hoshii T, Maekawa H,
Ogawa H, Suda T, Araki K and Yamamura K: Increase of smooth muscle
cell migration and of intimal hyperplasia in mice lacking the
alpha/beta hydrolase domain containing 2 gene. Biochem Biophys Res
Commun. 329:296–304. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Laemmli UK: Cleavage of structural
proteins during the assembly of the head of bacteriophage T4.
Nature. 227:680–685. 1970. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kyhse-Andersen J: Electroblotting of
multiple gels: a simple apparatus without buffer tank for rapid
transfer of proteins from polyacrylamide to nitrocellulose. J
Biochem Biophys Methods. 10:203–209. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Falk W, Goodwin RH Jr and Leonard EJ: A
48-well micro chemotaxis assembly for rapid and accurate
measurement of leukocyte migration. J Immunol Methods. 33:239–247.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nishiura H, Tokita K, Li Y, Harada K,
Woodruff TM, Taylor SM, Nsiama TK, Nishino N and Yamamoto T: The
role of the ribosomal protein S19 C-terminus in Gi
protein-dependent alternative activation of p38 MAP kinase via the
C5a receptor in HMC-1 cells. Apoptosis. 15:966–981. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Doran AC, Meller N and McNamara CA: Role
of smooth muscle cells in the initiation and early progression of
atherosclerosis. Arterioscler Thromb Vasc Biol. 28:812–819. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hugli TE: Biochemistry and biology of
anaphylatoxins. Complement. 3:111–127. 1986.PubMed/NCBI
|
|
24
|
Manthey HD, Woodruff TM, Taylor SM and
Monk PN: Complement component 5a (C5a). Int J Biochem Cell Biol.
41:2114–2117. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Naik N, Giannini E, Brouchon L and Boulay
F: Internalization and recycling of the C5a anaphylatoxin receptor:
evidence that the agonist-mediated internalization is modulated by
phosphorylation of the C-terminal domain. J Cell Sci.
110:2381–2390. 1997.PubMed/NCBI
|
|
26
|
Matsubara S, Yamamoto T, Tsuruta T, Takagi
K and Kambara T: Complement C4-derived monocyte-directed chemotaxis
inhibitory factor. A molecular mechanism to cause polymorphonuclear
leukocyte-predominant infiltration in rheumatoid arthritis synovial
cavities. Am J Pathol. 138:1279–1291. 1991.
|
|
27
|
Rua-Figueroa I, Arencibia-Mireles O,
Elvira M, Erausquin C, Ojeda S, Francisco F, Naranjo A,
Rodriguez-Gallego C, Garcia-Laorden I, Rodríguez-Perez J and
Rodríguez-Lozano C: Factors involved in the progress of preclinical
atherosclerosis associated with systemic lupus erythematosus: a
2-year longitudinal study. Ann Rheum Dis. 69:1136–1139.
2010.PubMed/NCBI
|
|
28
|
Monk PN, Scola AM, Madala P and Fairlie
DP: Function, structure and therapeutic potential of complement C5a
receptors. Br J Pharmacol. 152:429–448. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Weaver DJ Jr, Reis ES, Pandey MK, Köhl G,
Harris N, Gerard C and Köhl J: C5a receptor-deficient dendritic
cells promote induction of Treg and Th17 cells. Eur J Immunol.
40:710–721. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Braun M, Pietsch P, Schrör K, Baumann G
and Felix SB: Cellular adhesion molecules on vascular smooth muscle
cells. Cardiovasc Res. 41:395–401. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shagdarsuren E, Djalali-Talab Y,
Aurrand-Lions M, Bidzhekov K, Liehn EA, Imhof BA, Weber C and
Zernecke A: Importance of junctional adhesion molecule-C for
neointimal hyperplasia and monocyte recruitment in
atherosclerosis-prone mice-brief report. Arterioscler Thromb Vasc
Biol. 29:1161–1163. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jones SA: Directing transition from innate
to acquired immunity: defining a role for IL-6. J Immunol.
175:3463–3468. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tanaka J, Tajima S, Asakawa K, Sakagami T,
Moriyama H, Takada T, Suzuki E and Narita I: Preventive effect of
irbesartan on bleomycin-induced lung injury in mice. Respir
Investig. 51:76–83. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lee GL, Chang YW, Wu JY, Wu ML, Wu KK, Yet
SF and Kuo CC: TLR 2 induces vascular smooth muscle cell migration
through cAMP response element-binding protein-mediated
interleukin-6 production. Arterioscler Thromb Vasc Biol.
32:2751–2760. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xing J, Peng K, Cao W, Lian X, Wang Q and
Wang X: Effects of total flavonoids from Dracocephalum moldavica on
the proliferation, migration, and adhesion molecule expression of
rat vascular smooth muscle cells induced by TNF-α. Pharm Biol.
51:74–83. 2013.PubMed/NCBI
|
|
36
|
Norgauer J, Dobos G, Kownatzki E, Dahinden
C, Burger R, Kupper R and Gierschik P: Complement fragment C3a
stimulates Ca2+influx in neutrophils via a
pertussis-toxin-sensitive G protein. Eur J Biochem. 217:289–294.
1993.PubMed/NCBI
|
|
37
|
Zhou W: The new face of anaphylatoxins in
immune regulation. Immunobiology. 217:225–234. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhou X, Yang W and Li J:
Ca2+-and protein kinase C-dependent signaling pathway
for nuclear factor-kappaB activation, inducible nitric-oxide
synthase expression, and tumor necrosis factor-alpha production in
lipopolysaccharide-stimulated rat peritoneal macrophages. J Biol
Chem. 281:31337–31347. 2006.PubMed/NCBI
|
|
39
|
Castro R, Sun XH, Liu XB, Martinez JR and
Zhang GH: Inhibition of Ca2+influx by surfactant in
NR8383 alveolar macrophages. Inflamm Res. 57:489–496. 2008.
|