Introduction
Bronchopulmonary dysplasia (BPD) is a common and
serious complication in premature infants born at a gestational age
of <29 weeks (1). Among low
birth weight infants (<1,500 g), the incidence of BPD approaches
43% (2), and ~50% of children with
BPD are rehospitalized due to respiratory distress during early
childhood, particularly in cases of concomitant respiratory
syncytial viral infection (3).
Long-term dysplastic diseases of the respiratory and nervous system
are associated with a diagnosis of BPD in infancy and can persist
into adolescence and adulthood, compromising the individual’s
quality of life and resulting in substantial medical costs
(4).
Clinical studies have identified several risk
factors associated with the occurrence of BPD in premature
neonates, including hyperoxia, ventilator-induced pulmonary injury
and antenatal infection (5,6).
These various factors are considered to act in a cumulative and
synergic manner, causing early inflammation and lung injury, and
leading to fibrosis and abnormal maturation processes (7). The mechanism underyling BPD remains
to be fully elucidated, however, oxidative stress is important in
the occurrence and development of BPD. Reactive oxygen species
(ROS) induce lung injury and are a primary contributor to the
pathogenesis of BPD (8,9). Our previous study demonstrated that
oxidative stress-induced lung injury occurred during the
development of BPD in a rat model of neonatal hyperoxia (10,11),
consistent with other previous studies (12,13).
ROS induces significant, variable DNA damage, which
can result in a loss of normal cellular functioning (14–16),
unless cells activate timely DNA repair pathways (17). Oxidative DNA damage is repaired
predominantly by the base excision repair (BER) pathways (15,16).
In mammalian cells, 8-oxoguanine DNA glycosylase 1 (OGG1) is
important in BER pathways (18,19).
Overexpression of OGG1 in pulmonary artery endothelial cells
reduces xanthine oxidase-induced mitochondrial DNA damage and cell
apoptosis (20). By contrast, OGG1
knockdown by small interfering RNA in pulmonary artery endothelial
cells, delays xanthine oxidase-induced DNA damage repair by
mitochondria and increases the rate of cell apoptosis (21).
A previous study has suggested that OGG1 functions
to antagonize oxidative DNA damage (22). However, few studies have examined
whether lung epithelial DNA damage occurs during the process of
hyperoxia-induced BPD in neonatals (23) or whether this damage is associated
with OGG1. The present study examined the association between DNA
damage in lung epithelial cells and OGG1 in a neonatal rat model of
hyperoxia-induced BPD.
Materials and methods
Animals and hyperoxia exposure
A total of 20 pregnant Wistar rats (200–220 g) were
purchased from the Center for Experimental Animals of China Medical
University (Shengyang, China). All animal procedures were reviewed
and approved by the Laboratory Animal Ethics Committee of China
Medical University. The pups (n=80) were delivered naturally at
full-term gestation (22 days). All of the rats were maintained in
pathogen-free conditions and housed in a temperature- and
humidity-controlled environment. They were all subjected to a 12 h
light/12 h dark cycle and were given ad libitum access to
food and water. The newborn rats from 12 litters were randomly
assigned to either a hyperoxia-exposed group (90% O2) or
a normoxia (21% O2) control group, from the day of
birth. Inhaled oxygen concentrations were measured continuously
using an oxygen analyzer equipped with a strip-chart recorder
(model 572; Servomex, Co., Norwood, MA, USA). Humidity levels were
maintained between 60 and 70%. Nursing rat dams were exchanged
every 24 h between the hyperoxic and normoxic chambers to avoid
oxygen toxicity and to provide equivalent nutrition to all pups.
The chambers were opened for 10 min each day to weigh the pubs and
for cage cleaning. Following 1, 2, 3, 5 or 7 days of exposure, the
pups were sacrificed by abdominal aorta disconnection under
intraperitoneal (i.p.) anesthesia (0.6 ml/100 mg 5% chloral
hydrate; Sigma-Aldrich, St. Louis, MO, USA) and the lungs were
harvested for subsequent analyses.
Neonatal rat alveolar epithelial type II
cells (AECII)
The isolation, purification and culture of neonatal
rat AECII cells were performed, as described previously (24–26).
Briefly, the lungs of neonatal rats were removed within 24 h of
birth, following anesthesia with 5% chloral hydrate (0.6 ml/100 mg;
i.p.). The lungs were placed in cold D-Hank’s balanced buffer
solution, and the trachea, main bronchus and hilar tissues were
dissected and discarded; the remaining tissue was rinsed twice in
D-Hank’s balanced buffer solution and cut into small pieces (~1
mm3). The lung tissue was incubated in 10 ml
phosphate-buffered saline (PBS; Sigma-Aldrich) supplemented with
0.25% trypsin (Merck Millipore, Darmstadt, Germany) and 0.02%
ethylenediaminetetraacetic acid, in a constant temperature water
bath at 37°C with agitation for 30 min, in order to digest the
tissue. The same volume of Dulbecco’s modified Eagle’s medium
(DMEM; HyClone, Logan, UT, USA) containing 10% fetal bovine serum
(FBS; HyClone) was added to terminate the digestion. The
dissassociated cells were then filtered through a 200-mesh cell
strainer, centrifuged at 71.5 × g for 5 min and the supernatant was
discarded. Subsequently, 0.1% type I collagenase (Gibco Life
Technologies, Carlsbad, CA, USA) in D-Hank’s balanced buffer
solution was added to the re-suspended cells, which were digested
for 20 min at 37°C. The digested cells were centrifuged, the
supernatant was discarded and DMEM supplemented with 10% FBS was
used to resuspend the cells. Fibroblasts were removed using the
differential attachment procedure (repeated twice, 50 min/time).
Unattached cells were transferred to 100 mm culture dishes coated
with rat immunoglobulin G(IgG) (Abcam, Hong Kong, China), and the
AECII cells were further purified, based on the IgG binding
properties of the cells. Typan blue staining (Sigma-Aldrich) showed
that >94% of purified AECII cells were viable. The purity of the
isolated AECII cells was determined by calculating the positive
perentage of immunofluorescence staining of surfactant protein
(specific marker of AECII cells) in the isolated cells. The
isolated AECII cells were cultured in DMEM containing 10% FBS, 100
U/ml penicillin and 100 mg/ml streptomycin (Life Technologies,
Rockville, MD, USA) at 37°C in an atmosphere containing 21%
O2 and 5% CO2. Following culture, the cell
density was adjusted to 2–3×106 cells/ml and a portion
of the purified cells (0.4 ml) were seeded onto glass coverslips
(15 mm × 15 mm; WHEATON, Millville, NJ, USA) for immunofluorescence
staining whilst others (2 ml) were seeded into Petri dishes (Thermo
Fisher Scientific, Waltham, MA, USA) for analysis by competitive
enzyme-linked immunosorbent assay (ELISA), comet assay, western
blotting or reverse transcription quantitative polymerase chain
reaction (RT-qPCR). The cells from each isolation were cultured
with 21% O2 and 5% CO2 in an incubator
(Thermo Fisher Scientific) for 24 h. The cultures were replaced
with fresh medium to remove unattached cells and the attached cells
were randomly divided into either a 90% O2/5%
CO2-exposed hyperoxia group or a 21% O2/5%
CO2-exposed normoxia control group. The cells were then
cultured for 12, 24, 48 or 72 h at 37°C in an incubator (Thermo
Fisher Scientific) and were subsequently collected for the
corresponding experiments. The purity of the AECII cells was
~90–95% following 1 day of culture. The AECII cells were identified
using three methods: Inverted phase contrast microscopy,
transmission electron microscopy (TEM) and surfactant protein-C
(SP-C) detection by immunofluorescence staining.
Detection of 8-hydroxy-2′-deoxyguanosine
(8-OHdG)
A competitive ELISA for 8-OHdG was performed using a
commercial 8-OHdG ELISA kit (Cayman Chemicals Co., Ann Arbour, MI,
USA), according to the manufacturer’s instructions. The DNA was
purified from the lung tissues and AECII cells using a Wizard
Genomic DNA Purification kit (Promega Corporation, Madison, WI,
USA) and the DNA purity was confirmed by measuring the A260:A280
ratio. Enzymatic digestion was performed using nuclease P1 (pH 5.3;
Sigma-Aldrich) at 50°C for 1 h and with alkaline phosphatase (pH
8.5; Sigma-Aldrich) at 37°C for 30 min. The samples were boiled for
10 min and placed on ice for 5 min. The DNA hydrolysates were
analyzed by ELISA, according to the manufacturer’s instructions.
The plates were read at a wavelength of 412 nm (Tecan Sunrise
Microplate reader; Tecan, Männedorf, Switzerland) and the level of
8-OHdG was determined for each sample from a standard curve.
Comet assay
A Comet Assay kit (Cell Biolabs, Inc., Beijing,
China) was used for single cell gel electrophoresis, according to
the manufacturer’s instructions. Briefly, 1×106 cells/ml
AECII cells were washed with PBS and the cell suspension was mixed
with liquified agarose at a 1:9 (v/v) ratio. A small aliquot of
this mixture (100 μl) was immediately transferred to
agarose-coated slides (Thermo Fisher Scientific) and lysed (Cell
Lysis Solution; Sigma-Aldrich) at 4°C in the dark for 2 h.
Following cell lysis, the slides were treated with alkaline
solution containing 0.6 mM Na-EDTA and 0.18 M NaOH (pH 13) at room
temperature in the dark for 30 min to unwind the double-stranded
DNA. The slides were electrophoresed (Biolab Comet-061; Beijing
Biolab Science and Technology Co., Ltd, Beijing, China) under
alkaline conditions at room temperature at 1 V/cm for 20 min. and
subsequently neutralized using 0.4 M Tris (pH 7.5; Sigma-Aldrich)
and fixed with absolute ethanol (100%). Following 10 min staining
with SYBR Green dye (200 μl; Biotium, Inc., Hayward, CA,
USA), images were captured by fluorescence microscopy using a Nikon
E2000 Microscope system and the accompanying software (Nikon
Corporation, Tokyo, Japan). Images were scored for comet assay
parameters, including tail length, olive tail moment and the
percentage of DNA in the tail, using Comet A v.1.0 image analysis
software (Cells Biolab, Inc., Beijing, China).
Immunofluorescence staining
The left lobes of the lungs were inflated using 4%
paraformaldehyde, soaked in 3% paraformaldehyde for 3 h at 4°C,
cryoprotected in 30% sucrose (Sigma-Aldrich) for 12 h at 4°C and
then frozen at −80°C. The frozen sections (8 μm thick) were
air-dried and washed three times with PBS. The AECII cells were
fixed using 4% paraformaldehyde (Sigma-Aldrich) for 30 min and were
washed three times with PBS. The sections were incubated with 0.3%
Triton X-100 (Sigma-Aldrich) for 5 min at room temperature and
washed three times with PBS. The sections were then blocked with 5%
goat serum for 1 h at room temperature and incubated with goat
polyclonal anti-OGG1 primary antibody (1:50; ab115841; Abcam)
overnight at 4°C. Sections incubated in the absence of the primary
antibodies were used as negative controls. The tissue sections were
washed three times with PBS and incubated with tetramethylrhodamine
isothiocyanate-conjugated mouse anti-goat immunoglobulin G
secondary antibody (1:100; sc-3916; Santa Cruz Biotechnology, Inc.,
Santa Cruz, CA, USA) for 1 h at 37°C. The sections were washed
three times with PBS and the nuclei were stained with
4′,6-diamidino-2-phenylindole dihydrochloride (1:2,000;
Sigma-Aldrich) for 2 min. The slides were subsequently washed three
times with PBS and images were captured using an MTC-600 confocal
laser scanning microscope (Bio-Rad Laboratories, Inc., Hercules,
CA, USA).
Western blotting
The lung tissues or AECII cells were homogenized in
radioimmunoprecipitation assay lysis buffer with
phenylmethanesulfonyl fluoride (Sigma-Aldrich). Following
centrifugation at 15,000 × g for 10 min at 4°C, the protein lysates
in the supernatant were quantified using a Bicinchoninic Acid
Protein Assay kit (Beyotime Institute of Biotechnology, Shanghai,
China). The proteins were loaded and separated through a 10%
SDS-polyacrylamide gel and were transferred onto polyvinylidene
fluoride membranes (Merck Milipore, Boston, MA, USA). The membranes
were blocked in 5% non-fat milk dissolved in Tris-buffered saline
(TBS) for 2 h at room temperature prior to incubation overnight at
4°C with anti-OGG1 (1:400; Abcam) and anti-β-actin primary
antibodies (1:1,000; Santa Cruz Biotechnology, Inc.). The membranes
were then washed in TBS-0.2% Tween 20 (TBST) and incubated with
horseradish peroxidase-conjugated secondary antibody at 37°C for 2
h. The membranes were washed in TBST, as previously. An enhanced
chemiluminescence detection kit (EMD Millipore, Billerica, MA, USA)
and a WD-9413B Gel Imaging system (Liuyi Instrument Factory,
Beijing, China) were used for chemiluminescence analysis and
imaging. The bands were quantified using ImageJ 1.45s software
(National Institutes of Health, Bethesda, MD, USA) and the optical
densities of all bands were normalized to those of β-actin.
RT-qPCR
Total RNA was purified from lung tissues or the
AECII cells using TRIzol reagent (Invitrogen Life Technologies,
Carlsbad, CA, USA), according to the manufacturer’s instructions.
The RNA samples were treated with 10 μl DNase for 30 min
(Takara Biotechnology Co., Dalian, China), according to the
manufacturer’s instructions. The RNA purities were confirmed by
measuring the A260:A280 ratio and an aliquot of total RNA (1
μg) per sample was reversed-transcribed to cDNA using a
PrimeScript RT reagent kit (Takara Biotechnology Co.), according to
the manufacturer’s instructions. RT-qPCR was performed on an ABI
PRISM 7900HT system (Applied Biosystems Life Technologies, Foster
City, CA, USA) using equal volumes of cDNA (2μl) with a SYBR
Premix Ex Taq II kit (Takara Biotechnology Co.), according to the
manufacturer’s instructions. PCR was performed using specific
primer pairs (Table 1), according
to the following program: 95°C for 30 sec; 40 cycles of 95°C for 5
sec and 60°C for 34 sec; 95°C for 15 sec; 60°C for 1 min and 95°C
for 15 sec. Melting curve analyses were performed for the amplified
genes and the specificity and integrity of the PCR products were
confirmed by the presence of a single peak. For determination of
the relative gene expression levels, the target mRNA were amplified
using the same procedure and the expression levels were calculated
relative to β-actin using the 2−ΔΔCt method, as
described previously (27). in the
Applied Biosystems User Bulletin #2 ‘Relative quantification of
gene expression’.
 | Table IPrimers used for reverse
transcription quantitative polymerase chain reaction. |
Table I
Primers used for reverse
transcription quantitative polymerase chain reaction.
| Gene | Forward
(5′–3′) | Reverse
(5′–3′) |
|---|
| OGG1 |
CTAAGAAGACAGAAGGCTAGGTAG |
TGACTTTGATTTGGGATGTTTGC |
| β-actin |
CGTGCGTGACATTAAAGAG |
TTGCCGATAGTGATGACCT |
Statistical analysis
Statistical analysis was performed using SPSS 17.0
software (SPSS, Inc., Chicago, IL, USA). Data were analyzed by
one-way analysis of variance with a Bonferroni post-hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
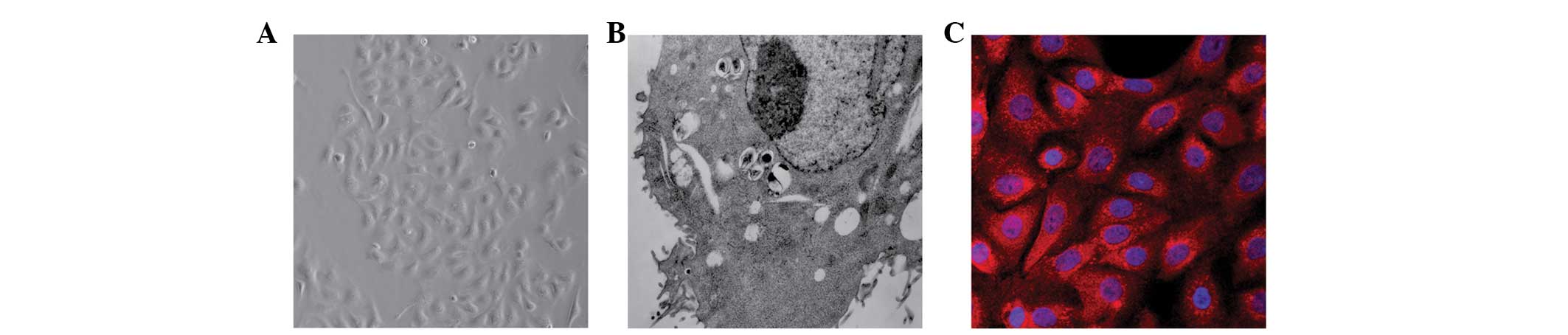
Identification of cultured neonatal rat
AECII cells
The cultured neonatal rat AECII cells were
identified using inverted phase contrast microscopy (Fig. 1A), TEM (Fig. 1B) and SP-C detection using
immunofluorescence staining (Fig.
1C).
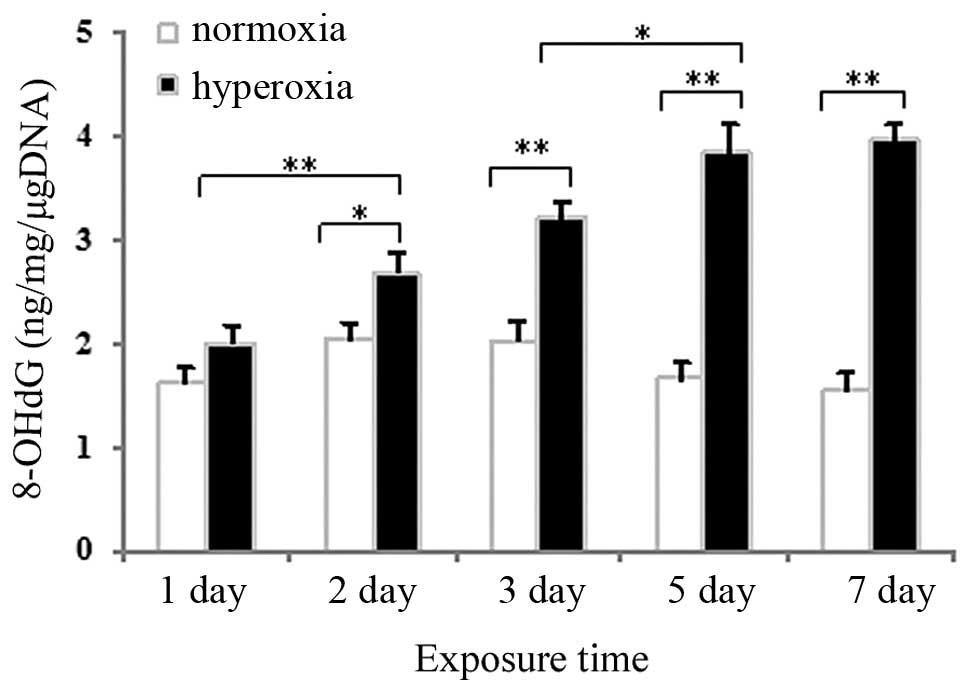
Effects of hyperoxia exposure on the
expression of 8-OHdG in neonatal rat lung tissues and cultured
AECII cells
Competitive ELISA was used to measure 8-OHdG, a
marker of oxidative DNA damage, in DNA samples from neonatal rat
lung tissues and in cultured AECII cells exposed to hyperoxia or
normoxia. As shown in Fig. 2, no
significant change in the expression of 8-OHdG was observed after 1
day of hyperoxia exposure, compared with the normoxia group
(P>0.05), whereas the expression of 8-OHdG in the lung tissues
exposed to hyperoxia for 2, 3, 5 or 7 days significantly increased
(P<0.05 for 2 days and P<0.01 for 3, 5 and 7 days). As shown
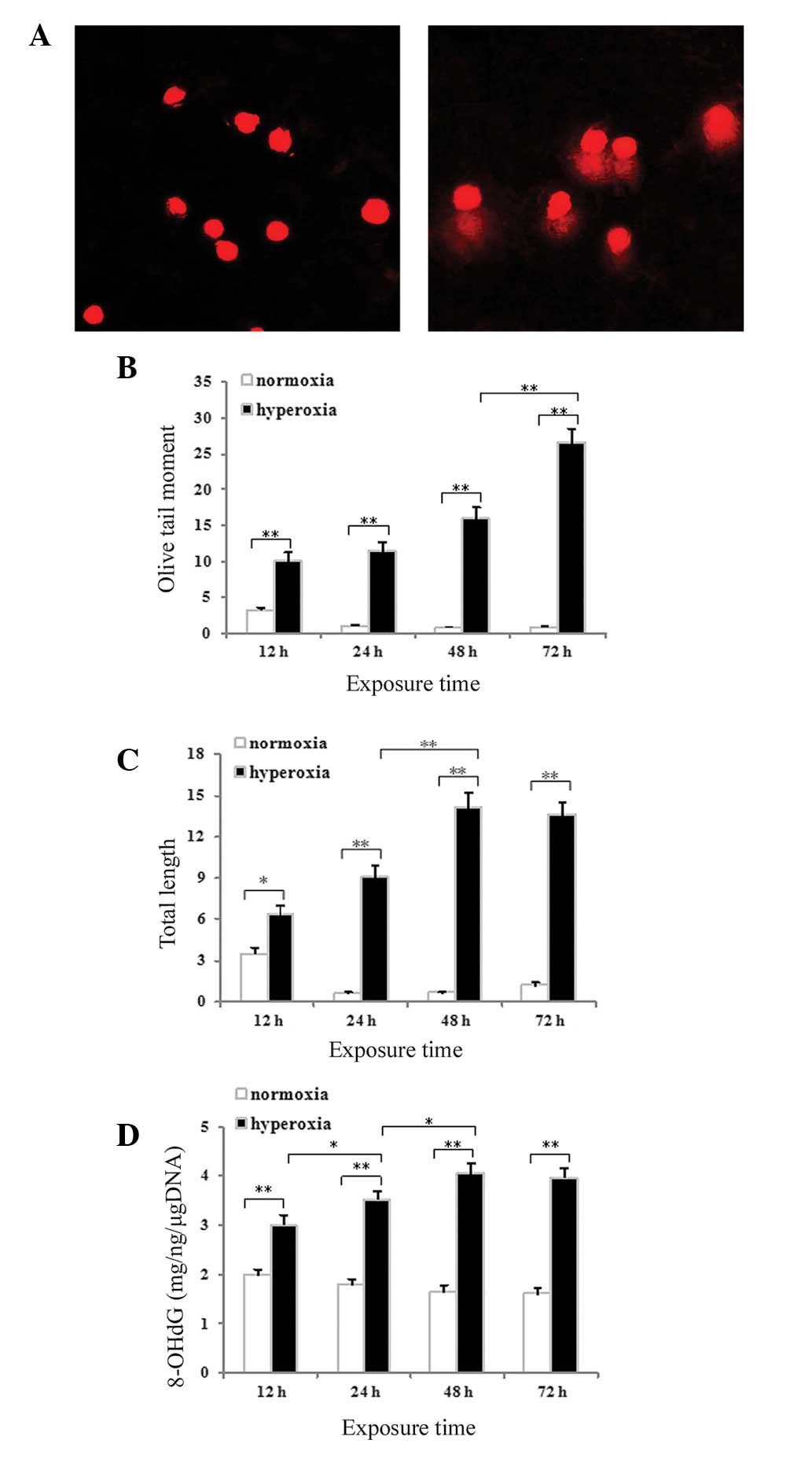
in Fig. 3E, hyperoxia exposure was
associated with significantly increased expression of 8-OHdG in the
AECII cells at all time-points compared with the normoxia control
(P<0.01) The 8-OHdG content in the lung tissues and cultured
AECII cells increased gradually as the hyperoxia exposure time
increased.
Hyperoxia-induced DNA strand breaks in
cultured neonatal rat AECII cells
An alkaline comet assay was used to assess DNA
strand breaks in the AECII cells. Minimal migration of DNA was
detected following exposure to normoxia for 48 h (Fig. 3A). By contrast, significant
migration of the DNA from the nucleus, forming a comet tail, was
observed following exposure to hyperoxia for 48 h (Fig. 3B). Measurements of comet tail
length and olive tail moment were used to evaluate the DNA strand
breaks. The tail length (P<0.05 for 12 h and P<0.01 for 24,
48 and 72 h; Fig. 3C) and the
olive tail moment (P<0.01 for 12, 24, 48 and 72 h; Fig. 3D) increased significantly with time
in the hyperoxia group compared with the normoxia control
group.
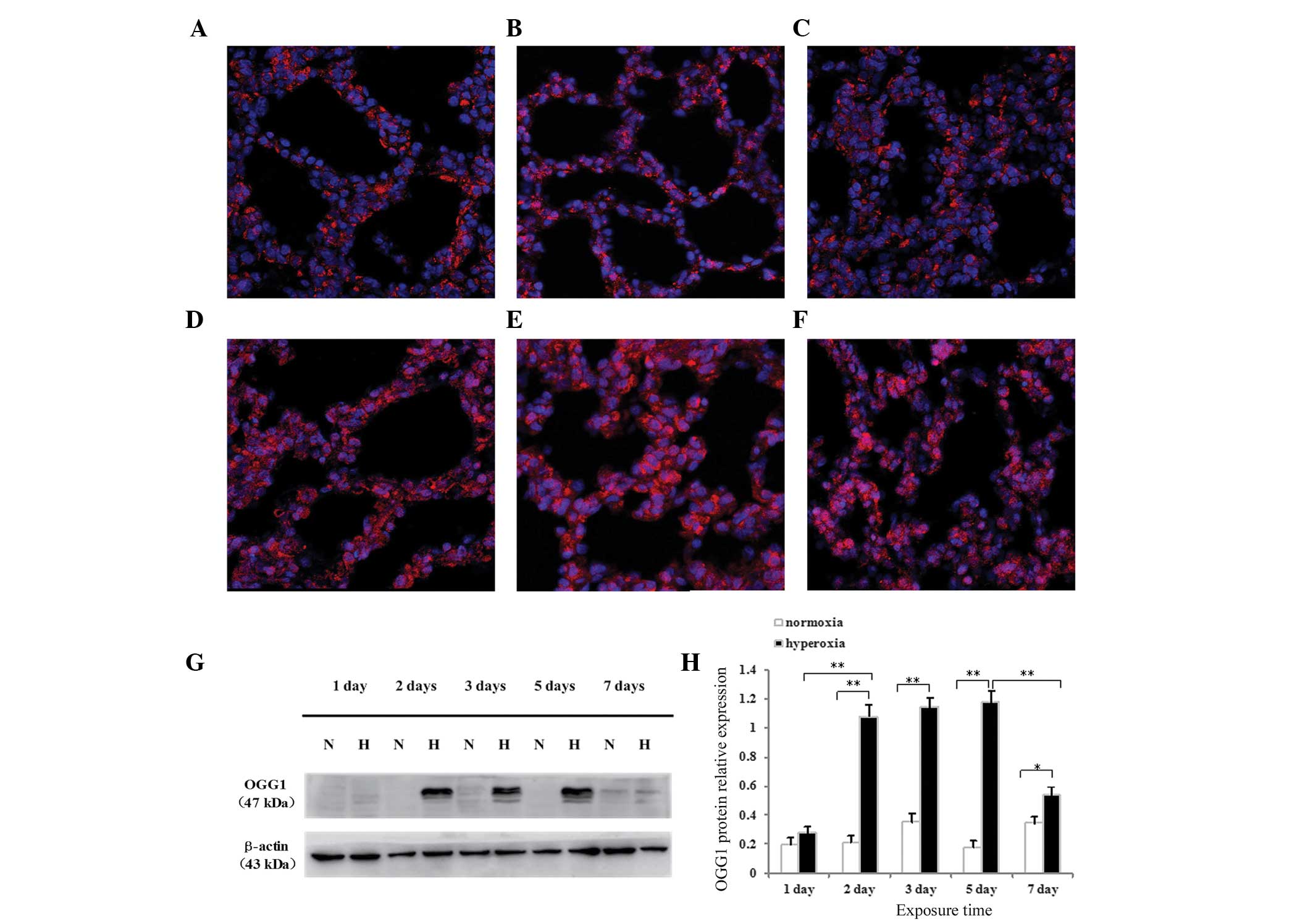
Effects of hyperoxia exposure on the
localization and expression of OGG1 protein in neonatal rat lung
tissues and cultured AECII cells
The expression and localization of OGG1 protein in
the alveolar epithelium of newborn rats were determined using
immunofluorescence confocal microscopy. Following normoxia exposure
for 1 day (Fig. 4A) or 5 days
(Fig. 4B), or hyperoxia exposure
for 1 day (Fig. 4C), OGG1 was
located primarily in the cytoplasm, and no difference was observed
between the hyperoxia and normoxia groups. Following hyperoxia
exposure for 3 days (Fig. 4D), 5
days (Fig. 4E) or 7 days (Fig. 4F), the localization of OGG1
significantly increased in the nucleus and, particularly in the
cytoplasm. The protein expression of OGG1 was highest following
hyperoxia exposure for 3 or 5 days. The protein expression of OGG1
was significantly reduced 7 days after hyperoxia, compared with 3
and 5 days, however, the expression remained elevated compared with
that observed on day 1.
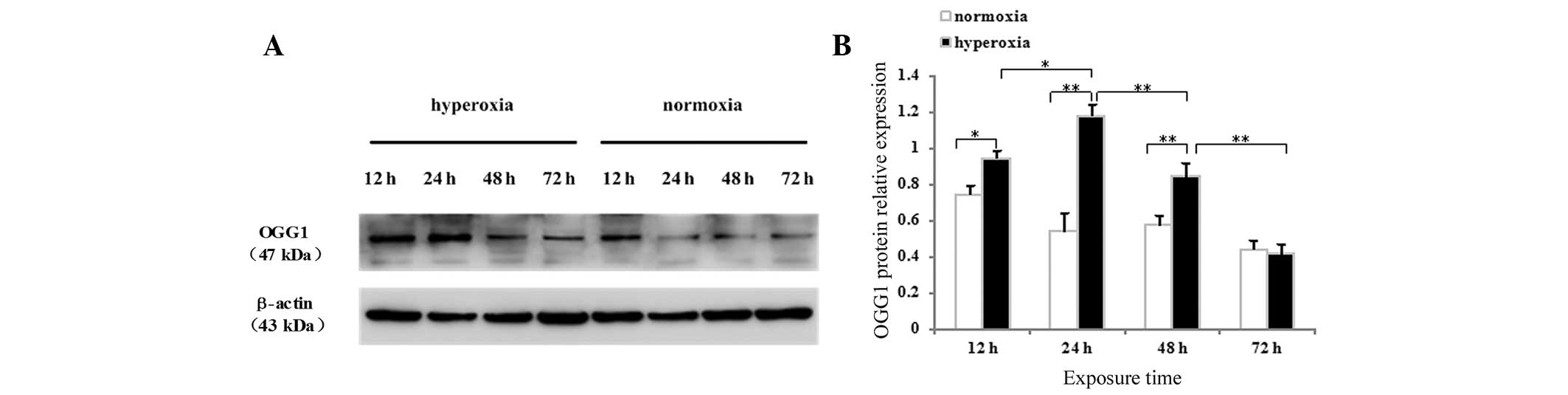
The present study also detected the protein
expression of OGG1 in neonatal rat lung tissues and cultured AECII
cells by western blotting. The OGG1 protein was identified as a
dominant band of ~47 kDa in the lung tissues (Fig. 4G) and AECII cells (Fig. 5A) exposed to hyperoxia or normoxia
at all time-points. As shown in Fig.
4F, no difference in the protein expression of OGG1 was
observed after 1 day of hyperoxia exposure compared with the
controls (P>0.05), whereas the protein expression of OGG1 in the
lung tissues exposed to hyperoxia for 2, 3, 5 or 7 days was
significantly increased (P<0.01 for 2, 3 and 5 days; P<0.05
for 7 days). The protein expression of OGG1 in the lung tissues
began to increase following 2 days of hyperoxia exposure, peaked
between 3 and 5 days and began to decrease following 7 days of
hyperoxia. As shown in Fig. 5B,
the protein expression of OGG1 increased significantly in cells
exposed to hyperoxia for 12, 24 and 48 h, compared with the
controls (P<0.05 for 12 h; P<0.01 for 24 and 48 h), whereas
no difference was observed in the protein expression of OGG1 72 h
after hyperoxia exposure (P>0.05). The protein expression of
OGG1 in the hyperoxia-exposed AECII cells began to increase after
12 h, peaked after 24 h, began to decrease after 48 h and decreased
further after 72 h.
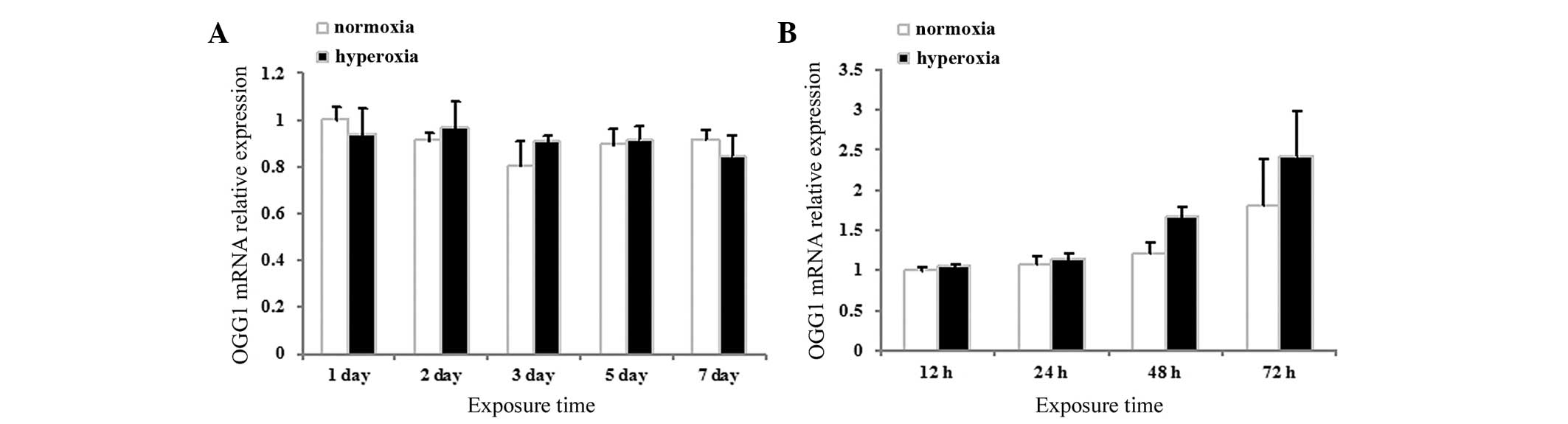
Effects of hyperoxia exposure on the mRNA
expression of OGG1 in neonatal rat lung tissues and neonatal rat
AECII cells
The mRNA expression levels of OGG1 in the neonatal
rat lung tissues and cultured AECII cells were assessed using
RT-qPCR. No significant differences were observed between the mRNA
expression levels of OGG1 in the hyperoxia and normoxia group in
either the lung tissues (P>0.05; Fig. 6A) or the cultured AECII cells
(P>0.05; Fig. 6B) at any of the
time-points.
Discussion
BPD is a multifactorial disease, however, oxidative
stress resulting from multiple causes is the predominant pathogenic
factor in BPD (28). Several
previous studies have confirmed that oxidative stress-induced lung
injury is involved in the occurrence and development of BPD
(29,30). The expression of macrophages and
interleukins are increased in the bronchial alveolar lavage fluid
of children with BPD (31,32) and animal models of BPD exhibited
increased levels of pro-inflammatory cytokines, including
interleukin-1 and tumor necrosis factor-α (33). Inhalation of high oxygen
concentrations can increase the lipid peroxidation of lung tissues
and cause oxidative stress damage in the lungs (10–13,32).
The importance of oxidative stress in BPD is well-established, and
the prevention and treatment of pulmonary oxidative damage have
been a focus of antioxidant therapies (34–36).
However, the effects of treating pediatric patients with BPD with
general antioxidants are insubstantial (34,37),
therefore, a novel therapeutic approach for the prevention and
treatment of BPD is imperative.
The most serious effect of oxidative stress is DNA
damage. In pre-term baboons, mechanical ventilation with high
concentrations of oxygen (100% O2) increased the level
of oxidative DNA damage to lung tissues compared with controls
receiving levels of oxygen required to maintain PO2
between 50 and 80 mmHg (38).
Marked oxidative DNA damage occurs following exposure of A549 cells
to 95% O2 and the damage increases gradually with
increasing exposure duration (39). The present study confirmed that the
occurrence of oxidative DNA damage was induced in the lung
epithelium of newborn rats by exposure to continuously high
concentrations of oxygen (90% O2). In addition, DNA
damage was exacerbated with increased hyperoxia exposure duration.
These findings are consistent with previous studies, demonstrating
that hyperoxia increases DNA damage (23,38,39).
ROS attack nuclear and mitochondrial DNA, inducing
various DNA mutations (40). BER
is regarded as the predominant DNA repair pathway, in which DNA
glycosylase-mediated identification and the excision of damaged
bases occurs (15,16). OGG1 is the most important BER
enzyme involved in this step (18,19).
In nuclear cataracts developed in adult Wistar rats exposed to 60%
oxygen, DNA damage in the lens increases concurrent with an
increased protein expression of OGG1 (41). Exposure of A549 cells to high
oxygen levels significantly increases the level of DNA damage and
overexpression of OGG1 in A549 cells, and adult rat AECII cells can
alleviate DNA damage and increase the survival rate of cells
exposed to hyperoxia (39). OGG1
is an established antagonist of DNA damage caused by oxidative
stress, however, the association between OGG1 and the development
of hyperoxia-induced BPD remains to be elucidated.
The present study used in vitro and in
vivo hyperoxia exposure experiments and confirmed that the
protein expression of OGG1 increased as the duration of hyperoxia
exposure increased during early-stage hyperoxia exposure, with an
increased in OGG1 in the cytoplasm. Following a peak in expression,
the protein expression of OGG1 decreased with increasing duration
of hyperoxia exposure. High oxygen potentially induced severe
oxidative DNA damage in the pulmonary epithelium, thereby
activating the DNA repair protein, OGG1. With extended periods of
hyperoxia exposure, the reduction in the protein expression of OGG1
may account for a concomitant suppression of DNA damage repair,
causing DNA damage to accumulate. The upregulation of OGG1 was
associated with an increased level of cytoplasmic OGG1. Therefore,
it was hypothesized that localization of OGG1 to the cytoplasm is
important in the occurrence of BPD. With its proximity to the
electron transport chain and the relatively limited mitochondrial
DNA repair capacity, mitochondrial DNA is significantly more
sensitive to ROS-mediated oxidative DNA damage, compared with
nuclear DNA (42,43). Previous studies have suggested that
mitochondrially localized OGG1 is involved in antagonizing the
oxidative DNA damage induced by ROS (20,21,44).
The in vitro and in vivo analyses performed in the
present study indicated no significant increase in the mRNA
expression of OGG1 at any time-point in the hyperoxia-exposed
groups. The mRNA expression of OGG1 was not consistent with the
protein expression of OGG1, therefore, the present study
hypothesized that OGG1 is regulated primarily at the level of
protein expression during the occurrence of hyperoxia-induced BPD.
The induction of oxidative DNA damage in the spleens of adult
Sprague Dawley rats exposed to subchronic aniline was associated
with significant increases in the protein and mRNA expression
levels of OGG1 (45), however,
this is not entirely consistent with the results of the present
study.
The present study demonstrated that severe DNA
damage occurred in lung epithelial cells during early-stage BPD.
The DNA repair gene, OGG1, may be important in this process and
further investigations are being performed to elucidate the
underlying regulatory mechanisms of OGG1 during hyperoxia-induced
BPD.
Acknowledgments
This study was supported by a grant from the Natural
Science Foundation of China (no. 30872781, 81170605).
References
|
1
|
Jobe AH and Bancalari E: Bronchopulmonary
dysplasia. Am J Respir Crit Care Med. 163:1723–1729. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bancalari E, Claure N and Sosenko IR:
Bronchopulmonary dysplasia: Changes in pathogenesis, epidemiology
and definition. Semin Neonatol. 8:63–71. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Greenough A, Cox S, Alexander J, et al:
Health care utilisation of infants with chronic lung disease,
related to hospitalisation for RSV infection. Arch Dis Child.
85:463–468. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Saigal S and Doyle LW: An overview of
mortality and sequelae of preterm birth from infancy to adulthood.
Lancet. 371:261–269. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Reyburn B, Martin RJ, Prakash YS, et al:
Mechanisms of injury to the preterm lung and airway: implications
for long-term pulmonary outcome. Neonatology. 101:345–352. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Van Marter LJ: Progress in discovery and
evaluation of treatments to prevent bronchopulmonary dysplasia.
Biol Neonate. 89:303–312. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Coalson JJ: Pathology of bronchopulmonary
dysplasia. Semin Perinatol. 30:179–184. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Northway WH Jr, Rosan RC and Porter DY:
Pulmonary disease following respirator therapy of hyaline-membrane
disease Bronchopulmonary dysplasia. N Engl J Med. 276:357–368.
1967. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gerschman R, Gilbert DL, Nye SW, et al:
Oxygen poisoning and x-irradiation: a mechanism in common. Science.
119:623–626. 1954. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jianhua F and Xindong X: Study on
oxidation and antioxidation in lung tissue of premature rat with
hyperoxia-induced chronic lung. Chin J Perinat Med. 7:361–363.
2004.
|
|
11
|
Jianhua Fu and Xindong Xue: Changes of
ultrastructure and oxidative stress reaction of lungs in premature
rats with chronic lung disease induced by hyperoxia. Chin J Contemp
Pediatrics. 6:23–26. 2004.
|
|
12
|
Cai Q and Xu MY: Protective effect of
rosiglitazone against hyperoxia-induced lung injury in neonatal
rats. Zhongguo Dang Dai Er Ke Za Zhi. 14:301–305. 2012.In Chinese.
PubMed/NCBI
|
|
13
|
Tayman C, Cekmez F, Kafa IM, et al:
Protective effects of nigella sativa oil in hyperoxia-induced lung
injury. Arch Bronconeumol. 49:15–21. 2013. View Article : Google Scholar
|
|
14
|
Jackson SP and Bartek J: The DNA-damage
response in human biology and disease. Nature. 461:1071–1078. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mitra S, Boldogh I, Izumi T, et al:
Complexities of the DNA base excision repair pathway for repair of
oxidative DNA damage. Environ Mol Mutagen. 38:180–190. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mitra S, Hazra TK, Roy R, et al:
Complexities of DNA base excision repair in mammalian cells. Mol
Cells. 7:305–312. 1997.PubMed/NCBI
|
|
17
|
Kruman II: Why do neurons enter the cell
cycle? Cell Cycle. 3:769–773. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hill JW, Hazra TK, Izumi T, et al:
Stimulation of human 8-oxoguanine-DNA glycosylase by
AP-endonuclease: potential coordination of the initial steps in
base excision repair. Nucleic Acids Res. 29:430–438. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vidal AE, Hickson ID, Boiteux S, et al:
Mechanism of stimulation of the DNA glycosylase activity of hOGG1
by the major human AP endonuclease: bypass of the AP lyase activity
step. Nucleic Acids Res. 29:1285–1292. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ruchko M, Gorodnya O, LeDoux SP, et al:
Mitochondrial DNA damage triggers mitochondrial dysfunction and
apoptosis in oxidant-challenged lung endothelial cells. Am J
Physiol Lung Cell Mol Physiol. 288:L530–L535. 2005. View Article : Google Scholar
|
|
21
|
Ruchko MV, Gorodnya OM, Zuleta A, et al:
The DNA glycosylase Ogg1 defends against oxidant-induced mtDNA
damage and apoptosis in pulmonary artery endothelial cells. Free
Radical Biol Med. 50:1107–1113. 2011. View Article : Google Scholar
|
|
22
|
Miller-Pinsler L and Wells PG: Deficient
DNA repair exacerbates ethanol-initiated DNA oxidation and
embryopathies in ogg1 knockout mice: gender risk and protection by
a free radical spin trapping agent. Arch Toxicol. Oct 30–2014.Epub
ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Auten RL, Whorton MH and Nicholas Mason S:
Blocking neutrophil influx reduces DNA damage in hyperoxia-exposed
newborn rat lung. Am J Respir Cell Mol Biol. 26:391–397. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang QY, Fu JH and Xue XD: Primary cell
culture and identification of alveolar epithelial cell isolated
from neonatal rats. Zhongguo Xinshengerke Zazhi. 25:339–341.
2010.
|
|
25
|
Li T, Koshy S and Folkesson HG:
Involvement of aENaC and Nedd4-2 in the conversion from lung fluid
secretion to fluid absorption at birth in the rat as assayed by RNA
interference analysis. Am J Physiol Lung Cell Mol Physiol.
293:L1069–L1078. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ji W, Fu J, Nie H, et al: Expression and
activity of epithelial sodium channel in hyperoxia-induced
bronchopulmonary dysplasia in neonatal rats. Pediatr Int.
54:735–742. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Motalleb G, Pourrahmat E, Najafi S, et al:
Epidermal growth factor receptor gene expression evaluation in
colorectal cancer patients. Indian J Cancer. 51:358–362. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Welty SE: Is there a role for antioxidant
therapy in bronchopulmonary dysplasia? J Nutr. 131:947S–950S.
2001.PubMed/NCBI
|
|
29
|
Sampath V, Garland JS, Helbing D, et al:
Antioxidant response genes sequence variants and BPD susceptibility
in VLBW infants. Pediatr Res. Dec 17–2014.Epub ahead of print.
PubMed/NCBI
|
|
30
|
Sandal G, Mutlu B, Uras N, et al:
Evaluation of treatment with hydrocortisone on oxidant/antioxidant
system in preterm infants with BPD. Eur Rev Med Pharmacol Sci.
17:2594–2597. 2013.PubMed/NCBI
|
|
31
|
Groneck P, Reuss D, Götze-Speer B, et al:
Effects of dexamethasone on chemotactic activity and inflammatory
mediators in tracheobronchial aspirates of preterm infants at risk
for chronic lung disease. J Pediatr. 122:938–944. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bracci R: Free oxygen radicals and
surfactant. Biol Neonate. 71:23–27. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Polglase GR, Dalton RG, Nitsos I, et al:
Pulmonary vascular and alveolar development in preterm lambs
chronically colonized with Ureaplasma parvum. Am J Physiol Lung
Cell Mol Physiol. 299:L232–L241. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Watts JL, Milner R, Zipursky A, et al:
Failure of supplementation with vitamin E to prevent
bronchopulmonary dysplasia in infants less than 1,500 g birth
weight. Eur Respir J. 4:188–190. 1991.PubMed/NCBI
|
|
35
|
Davis JM, Rosenfeld WN, Richter SE, et al:
Safety and pharmacokinetics of multiple doses of recombinant human
CuZn superoxide dismutase administered intratracheally to premature
neonates with respiratory distress syndrome. Pediatrics. 100:24–30.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ozdemir R, Yurttutan S, Talim B, et al:
Colchicine protects against hyperoxic lung injury in neonatal rats.
Neonatology. 102:265–269. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ahola T, Lapatto R, Raivio KO, et al:
N-acetylcysteine does not prevent bronchopulmonary dysplasia in
immature infants: a randomized controlled trial. J Pediatr.
143:713–719. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Maniscalco WM, Watkins RH, Roper JM, et
al: Hyperoxic ventilated premature baboons have increased p53,
oxidant DNA damage and decreased VEGF expression. Pediatr Res.
58:549–556. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kannan S, Pang H, Foster DC, et al: Human
8-oxoguanine DNA glycosylase increases resistance to hyperoxic
cytotoxicity in lung epithelial cells and involvement with altered
MAPK activity. Cell Death Differ. 13:311–323. 2006. View Article : Google Scholar
|
|
40
|
Cooke MS, Evans MD, Dizdaroglu M, et al:
Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J.
17:1195–1214. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang Y, Ouyang S, Zhang L, et al:
Oxygen-induced changes in mitochondrial DNA and DNA repair enzymes
in aging rat lens. Mech Ageing Dev. 131:666–673. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bohr VA, Stevnsner T and de Souza-Pinto
NC: Mitochondrial DNA repair of oxidative damage in mammalian
cells. Gene. 286:127–134. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yakes FM and Van Houten B: Mitochondrial
DNA damage is more extensive and persists longer than nuclear DNA
damage in human cells following oxidative stress. Proc Natl Acad
Sci USA. 94:514–519. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
He YH, Xu Y, Kobune M, et al: Escherichia
coli FPG and human OGG1 reduce DNA damage and cytotoxicity by BCNU
in human lung cells. Am J Physiol Lung Cell Mol Physiol.
282:L50–L55. 2002. View Article : Google Scholar
|
|
45
|
Ma H, Wang J, Abdel-Rahman SZ, et al:
Oxidative DNA damage and its repair in rat spleen following
subchronic exposure to aniline. Toxicol Appl Pharmacol.
233:247–253. 2008. View Article : Google Scholar : PubMed/NCBI
|