With the increasing morbidity of coronary heart
disease and hypertension, the prevalence of chronic heart failure
(CHF) has been increasing over the past several decades (1). CHF is the terminal state of various
cardiovascular diseases and the mortality rate remains high in
spite of the large number of studies which have been performed to
clarify its underlying mechanisms and to improve its treatment
(2). The most significant hallmark
of CHF is the continuous interaction between the underlying
myocardial dysfunction and the compensatory neurohumoral mechanisms
(3). Activation of the sympathetic
nervous system (SNS) is a major compensatory mechanism in the
development of CHF, which may be due to the changes in peripheral
baroreceptor and chemoreceptor reflexes, chemical mediators that
control sympathetic outflow and central integrative sites (4). Recent studies have indicated that a
variety of agents, including angiotensin II (AngII), nitric oxide
(NO) and pro-inflammatory cytokines are involved in the regulation
of sympathetic nerve activity (SNA) in the central nervous system
(CNS) during CHF (3). The
interaction between these agents and neurotransmitters can
influence the neuronal activity via central autonomic pathways in
different autonomic regions in the CNS. The abnormal
sympathoexcitation in the CNS deteriorates the cardiac function
during CHF and contributes to disease progression.
The paraventricular nucleus (PVN) in the
hypothalamus, the rostral ventrolateral medulla (RVLM) and the
nucleus tractus solitarius (NTS) are three important regions within
the CNS controlling SNA (5). The
NTS receives direct input from the cardiopulmonary afferents, such
as baroreceptors and chemo-receptors, and the area postrema (AP),
and has an indirect effect on the neuronal activity of the RVLM
through modulating the caudal ventrolateral medulla (CVLM) region
(6,7). In addition, the NTS has neural
connections to the PVN, a major integrative nucleus that can
influence SNA and extracellular fluid volume, and the
intermediolateral (IML) column of the spinal cord (5,8). The
PVN receives signals from the sub-fornical organ (SFO), which
positively influences the activity of the PVN (9). The RVLM has a key role in determining
the tonic and reflex control of SNA (10). It receives input signals from the
PVN and subsequently communicates with the IML, which is also under
direct influence of the PVN (11–13).
Finally, the IML receives input signals from these sympathetic
activity-regulating nuclei and stimulates the sympathetic
ganglionic neurons projecting to target organs.
Previous studies have demonstrated that the
renin-angiotensin system (RAS), whose main effective molecule is
AngII, is a key regulator of cardiovascular system function and
fluid homeostasis. RAS is activated upon decreases and inhibited
upon increases of the blood pressure, increases of sodium delivery
to the macula densa and volume overload. AngII has two types of
receptor: AngII type 1 (AT1) receptor is responsible for water and
sodium retention, aldosterone secretion and vasoconstriction, while
AT2 receptor is able to dilate the blood vessels.
According to the classic mechanism, AngII exerts its
effects via the circulating system. For example, the concentration
of AngII in plasma is increased under CHF conditions, which can
inhibit the sympathoinhibitory baroreflex and enhance the
sympathoexcitatory chemoreflex (14,15).
Apart from these actions, circulating AngII can also increase SNA
in the CNS through binding to its AT1 receptors in certain
autonomic regions, which have no blood-brain barrier, such as AP
and SFO (16,17). However, a large number of studies
have demonstrated the presence of an endogenous brain RAS within
the CNS (18). This is supported
by the fact that angiotensinogen, renin and angiotensin-converting
enzyme (ACE) are generated by glial cells and neurons in a number
of nuclei (19–21).
Microinjection of AngII into the PVN was shown to
increase plasma norepinephrine, renal sympathetic nerve activity
(RSNA), mean arterial pressure (MAP) and the heart rate (HR) to a
greater extent in rats with ischemia-induced CHF compared with
those in sham rats, whereas inhibition of AT1 receptors produced a
significant decrease of RSNA, MAP and HR (22). Administration of AngII into the
RVLM evoked sympathoexcitation in sham rats as well as rats with
CHF, with a significantly larger response in the animals with CHF
(23). In addition, basal SNA and
the cardiac sympathetic afferent reflex were shown to be decreased
by bilateral NTS injection of the AT1 receptor antagonist losartan
in rats with CHF but not in sham rats, suggesting that activation
of the AT1 receptor in the NTS can increase SNA during CHF
(21). Over-activity of the RAS of
the endogenous brain may be partly attributed to the increased
expression of AT1 receptor and ACE in the CNS. In CHF, the activity
of ACE in the PVN is increased and the expression of AT1 receptor
is also increased in the neurons of the PVN, RVLM and NTS (23–26).
Further investigation showed that AngII-triggered mitogen-activated
protein kinases (MAPKs) have an important role in the upregulation
of AT1 receptor in the PVN in rats with CHF (26). Under normal conditions, AT1
receptor is weakly expressed in astrocytes, while it is upregulated
in brainstem astrocytes and has a key role in enhancing sympathetic
outflow under CHF conditions. However, the expression of AT1
receptor in astrocytes of other sympathetic activity-regulating
nuclei during CHF requires further study (27). In an ovine model of CHF, central
infusion of losartan significantly decreased the elevated levels of
cardiac sympathetic nerve activity (CSNA), while RSNA was neither
elevated nor affected by losartan. Furthermore, the expression of
AT1 receptor was increased in the PVN but decreased in the NTS
(28). These results were not
completely consistent with the observations made in rodent models
of CHF and the reasons for these discrepancies remain elusive. In
spite of these discrepancies, the abovementioned studies suggested
that increased AngII signaling, via activating the AT1 receptor, is
responsible for the enhanced SNA during CHF.
In contrast to the AT1 receptor, the AT2 receptor
has an opposite role in regulating SNA within the CNS.
Intracerebroventricular injection of AngII in AT2 receptor gene
knock-out mice resulted in a greater increase in blood pressure
compared to that in wild-type mice (29). Intracerebroventricular infusion of
Compound 21, a non-peptide AT2 receptor agonist, decreased
norepinephrine excretion and blood pressure via the NO signaling
pathway in the PVN of normal rats and Compound 21 also suppressed
the sympathetic outflow by improving baroreflex sensitivity in rats
with CHF (30,31). Overexpression of AT2 receptor in
the RVLM led to a significant decrease in MAP and urinary
noradrenaline excretion in normal rats (32). However, the expression of AT2
receptor in the RVLM of CHF rats was significantly lower than that
in sham rats. These studies indicated that AT2 receptor activation
within the CNS causes a decrease in SNA and that impairment of AT2
receptor signaling is involved in the sympathoexcitation in CHF
(23). While the exact underlying
mechanisms for the beneficial effects of AT2 receptor activation
have yet to be elucidated, it has been indicated that activation of
the AT2 receptor may inhibit the neuronal excitability and decrease
the sympathetic outflow through increasing the potassium current in
neurons (33). However, direct
evidence for the sympathoinhibitory effects of AT2 receptor
activation in the NTS and PVN on the modulation of SNA during CHF
is currently lacking and the mechanisms of AT2 receptor
downregulation in CHF state require further investigation.
Glutamate and γ-aminobutyric acid (GABA) were
observed to have a role in the regulation of SNA within the CNS.
The PVN contains excitatory and inhibitory neurons, which use
glutamate and GABA as neurotransmitters. Microinjection of AngII
into the PVN resulted in an increase in glutamatergic and a
decrease in GABAergic transmission (34,35).
Glutamatergic neurons also exist in the RVLM and project to the
IML. Microinjection of AngII into the RVLM increased the release of
glutamate in the IML, which then enhanced the sympathetic outflow
to target organs (36). In
addition, microinjection of AngII into the NTS potentiated GABA
release through the effect of endothelial-derived NO. Elevated
levels of GABA inhibit the activity of glutamatergic neurons within
the NTS, which can further inhibit the GABAergic neurons in the
CVLM. As the GABAergic neurons in the CVLM have an inhibitory
effect on the glutamatergic neurons in the RVLM, the activity of
these glutamatergic neurons subsequently increases and potentiates
the sympathetic outflow (2,37,38).
Thus, the interaction between AngII, glutamate and GABA has an
important role in controlling SNA.
ROS, which include oxygen ions, free radicals and
peroxides, are generated during the normal metabolism of enzymes
such as NADPH oxidase. They are converted by superoxide dismutase
(SOD) into hydrogen peroxide and then rapidly degraded by enzymes.
Accumulating evidence indicated that ROS are important mediators of
AngII signaling within the CNS and are involved in the regulation
of SNA (39–41). Intracerebroventricular infusion of
AngII significantly increased RSNA in rabbits with CHF accompanied
by increased levels of NADPH-dependent ROS in the RVLM. In
addition, pre-treatment with an intra-cerebroventricular infusion
of SOD mimetic tempol and the inhibitor of NADPH oxidase apocynin
abolished the increased RSNA induced by AngII, suggesting that ROS
mediates the sympathoexcitation of AngII (42). Although the exact mechanisms by
which AngII induces ROS generation have remained elusive, it has
been reported that application of AngII caused NADPH-dependent ROS
production in NTS neurons, which was dependent on intracellular
Ca2+ and protein kinase C (43). ROS also have an important role in
AngII-induced AT1 receptor upregulation in CHF.
Intracerebroventricular infusion of tempol significantly decreased
AT1 receptor protein expression in the RVLM of rabbits with CHF;
furthermore, tempol reversed the AngII-induced increases in AT1
receptor mRNA expression in a neuronal cell line through inhibition
of the c-Jun N-terminal kinase and AP1 signaling pathways (44). Application of AngII decreases the
GABAergic currents in PVN neurons, which can be abolished by
treatment with SOD; in addition, ROS enhances excitatory inputs
from the PVN to the RVLM through increasing glutamatergic and
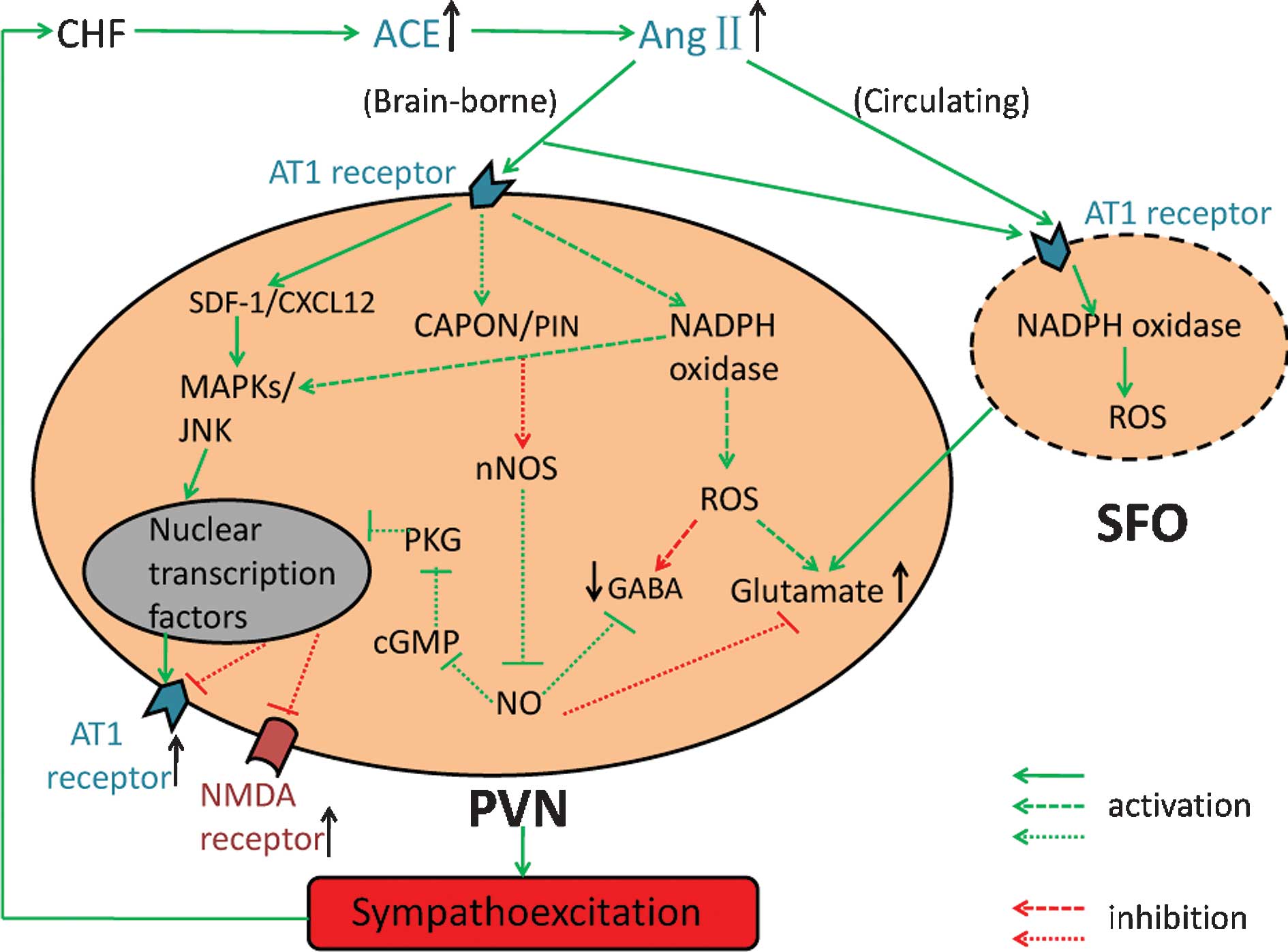
decreasing GABAergic transmission (Fig. 1) (35,45).
Furthermore, physical exercise decreased SNA via enhancing
anti-oxidant pathways and suppressing pro-oxidant mechanisms in the
RVLM of rabbits with CHF (46).
These studies indicated that NADPH-dependent ROS are involved in
the AngII signaling pathway and contribute to the increased
sympathetic outflow.
NO, a metabolic product of the arginine metabolism
catalyzed by nitric oxide synthase (NOS), is recognized as another
modulator of SNA in the CNS. NOS occurs in three isoforms: Neuronal
NOS (nNOS), endothelial NOS (eNOS) and inducible NOS (iNOS). nNOS
has been found to be involved in the control of sympathetic outflow
within all nuclei in the PVN, NTS and RVLM, while eNOS and iNOS are
mainly expressed in neurons in the brainstem (47–50).
Most of the previous studies demonstrated that NO generated by
these NOS isoforms exerts inhibitory effects on SNA (51).
As mentioned above, the baroreflex is impaired in
CHF, which contributes to sympathoexcitation. Gene transfer of nNOS
into the RVLM was shown to normalize the impaired baro-reflex
function (52). Similarly,
overexpression of eNOS in the NTS reduced urinary norepinephrine
excretion in mice with CHF (53).
Microinjection of nNOS anti-sense into the PVN elicited a
significant pressor in normal rats, while rats with CHF were less
affected, suggesting that the sympathoexcitatory effect of nNOS
anti-sense is blunted in CHF. This effect of nNOS anti-sense was
accompanied by decreased nNOS protein levels in the PVN and a
decreased responsiveness to NO in animals with CHF (54,55).
The expression of nNOS protein was demonstrated to be downregulated
in the NTS and the RVLM in rats with CHF (56). Chronic administration of AT1
receptor antagonist losartan prevented the downregulation of nNOS
gene expression in the PVN, NTS and RVLM during CHF (57). Thus, it is indicated that enhanced
AngII signaling in CHF decreases the expression of nNOS; however,
the underlying mechanisms remain elusive. It has been demonstrated
that the expression of carboxy-terminal PDZ ligand of nNOS (CAPON),
a protein which can interact with nNOS and prevent NO production
(58), was increased, whereas the
expression of nNOS was decreased in the PVN of rats with CHF
(59). Furthermore, treatment of
losartan abrogated the changes in expression of CAPON and nNOS in
animals with CHF, but also abolished AngII-induced increases in
CAPON in neuronal cells (59).
These results indicated that CAPON may act as a downstream mediator
in AngII-induced downregulation of nNOS during CHF (59). In addition, the expression of
protein inhibitor of nNOS (PIN) is upregulated due to increased
AngII levels in the PVN under CHF conditions. Binding of PIN to
nNOS leads to the de-stabilization of nNOS. As a result, the
inactive monomeric state of nNOS is susceptible to ubiquitination
and proteasomal degradation (Fig.
1) (60). These results
suggested that the sympathoinhibitory effect of NO is impaired by
decreases of nNOS, which thereby contribute to sympathoexcitation
during CHF.
Excitatory and inhibitory neurotransmitters have
been demonstrated to be involved in the mechanism by which NO
regulates the SNA. Administration of NO into the PVN has been shown
to increase local levels of GABA (61). NO-induced decreases in RSNA, blood
pressure and HR were inhibited by blocking of the GABAergic system,
which indicated that the inhibitory effects of NO within the PVN
are mediated by GABA (62).
However, administration of N-methyl-d-aspartate (NMDA) into the PVN
increased RSNA, blood pressure and HR. These responses were
enhanced by prior microinjection of NOS inhibitor
NG-monomethyl-l-arginine (L-NMMA), indicating
that NO inhibits NMDA-mediated increases in SNA in the PVN
(63). Gene transfer of nNOS into
the PVN of rats with CHF significantly enhanced the blunted changes
in RSNA, blood pressure and HR in response to L-NMMA, while the
response to NMDA was significantly decreased. This effect was due
to gene transfer of nNOS reducing the increased NMDA receptor
sub-unit NR(1) mRNA and protein
expression in the PVN of rats with CHF (64). Similarly, gene transfer of eNOS
into the RVLM significantly decreased MAP and HR in normal rats,
coupled with an increased expression of GABA. By contrast,
microinjection of GABA receptor antagonist following gene transfer
enhanced the increases in MAP (65). In addition, inhibition of iNOS in
the RVLM enhanced the pressor response caused by glutamate,
suggesting that NO derived from iNOS inhibited the action of
glutamate in the RVLM (66). In
vitro, NO negatively regulated AT1 receptor expression via the
protein kinase G pathway in primary cultures of hypothalamic and
neuronal cell lines (67),
indicating that reduced NO production within the CNS during CHF may
enhance the activity of the RAS (Fig.
1).
However, due to conflicting results, no consensus
has been reached regarding the role of NO in the regulation of SNA
in the RVLM and NTS. It has been reported that nNOS inhibition in
the RVLM decreased MAP and the HR. Microinjection of nNOS inhibitor
attenuated the pressor response caused by glutamate, indicating
that NO derived from nNOS produces sympathoexcitation through
enhancing the effects of glutamate in the RVLM (66). In addition, microinjection of
L-NMMA or an nNOS inhibitor into the RVLM attenuated the cardiac
sympathetic afferent reflex elicited by epicardial application of
bradykinin (68). In the NTS, NO
was shown to be able to potentiate GABAergic as well as
glutamatergic transmission (69).
However, the activation of glutamatergic transmission in the NTS
evoked a decrease in blood pressure and HR (70).
It is therefore indicated that through interacting
with RAS and neurotransmitters, NO produced by various isoforms of
NOS is able to decrease the SNA, while the sympathoinhibitory
effect of NO is impaired in CHF. Of note, in the RVLM and NTS, NO
can also increase SNA. Recently, it has been reported that
endogenous NO in the CNS exerted excitatory effects to increase
resting CSNA in healthy subjects, while exogenously administrated
NO inhibited CSNA under normal and CHF conditions (71). Thus, the sympathoexcitatory and
sympathoinhibitory effects of NO in the CNS may partly depend on
the balance of glutamatergic and GABAergic activity, the specific
sympathetic activity-regulating nuclei on which it acts and the
method of NO administration.
Pro-inflammatory cytokines, including tumor necrosis
factor-α (TNF-α) and interleukin-6 (IL-6), are elevated in CHF and
contribute to the progression of the disease (72). In analogy to AngII, peripheral
pro-inflammatory cytokines can penetrate into the CNS at the nuclei
where no blood-brain barrier exists; furthermore, cytokines can
also be generated by multiple cell types in the CNS (73,74).
Pro-inflammatory cytokines within the CNS can increase the
sympathetic outflow during CHF (75).
Through either acting on the SFO to produce multiple
neurohumoral factors or activating the perivascular macrophages to
generate prostaglandin E2 (PGE2), circulating pro-inflammatory
cytokines can further stimulate the activity of the PVN and
increase the sympathetic outflow (74,76).
The central inflammation-induced PGE2 generation depends on the
NADPH-oxidase and the p38 MAPK pathway under pathophysiological
conditions (77). Furthermore,
activated cardiac sympathetic afferent nerves transmit signals to
the CNS to increase cytokine production, which contributes to
sympathoexcitation by upregulation of the activity of the RAS and
the hypothalamic-pituitary-adrenal axis in the PVN under CHF
conditions (78,79). It has been indicated that nuclear
factor (NF)-κB may mediate the interaction between the RAS and
pro-inflammatory cytokines, as inhibition of the synthesis of NF-κB
reduced cytokine and AT1 receptor expression in the PVN of rats
with CHF and in turn, blockade of the AT1 receptor decreased the
expression of cytokines and NF-κB (80). In addition, central TNF increased
the NADPH oxidase sub-unit and ROS production in the PVN and RVLM
under CHF conditions, which further activated the RAS and NF-κB
(81,82). Toll-like receptor (TLR) signaling
has an important role in mediating the inflammation cascade.
Stimulation of TLR4 led to the activation of NF-κB via the myeloid
differentiation primary-response protein 88 (MyD88)-dependent
pathway, which induced pro-inflammatory cytokine production
(83). Intracerebroventricular
infusion of an AT1 receptor blocker decreased the elevated SNA and
reduced the increased expression of TLR4, MyD88 and NF-κB in the
brainstem of mice with CHF. Therefore, it is indicated that TLR4 is
involved in AT1 receptor-induced pro-inflammatory cytokine
production in CHF (84). Cytokines
were also indicated to upregulate SNA via increasing excitatory
neurotransmitters and decreasing inhibitory neurotransmitters, as
intracerebroventricular infusion of cytokine blockers attenuated
HF-induced increases in glutamate and decreases in GABA in the PVN
of rats with CHF, probably through an NO-associated mechanism
(Fig. 2) (85). In addition, chemokine stromal
cell-derived factor-1 may mediate the sympathoexcitation of TNF-α
and AngII in the PVN through activating p44/42 MAPK signaling,
which further activates the transcription factors and upregulates
the expression of AT1 receptor (Fig.
1) (86).
CHF is a clinical syndrome characterized by
continuous interaction between the underlying myocardial
dysfunction and compensatory neurohumoral mechanisms. Activation of
neurohumoral mechanisms in the CNS contributes to the
sympathoexcitation in CHF. Numerous studies have explored the role
of AngII, NO and pro-inflammatory cytokines in the regulation of
SNA in the CNS. By acting on sympathetic activity-regulating nuclei
at various levels, these neurohumoral factors influence the
activity of neurons and finally produce a sympathoexcitatory or
sympathoinhibitory effect. However, the underlying mechanisms by
which these factors regulate SNA remain to be fully elucidated and
the interaction between these agents within the CNS also remains to
be clarified. Finally, although blockade of the abnormal
neurohumoral axis in the CNS has shown beneficial effects in animal
experiments, further study is required to determine whether the
central neurohumoral axis may represent a novel target for the
treatment of patients with CHF.
|
1
|
Azad N and Lemay G: Management of chronic
heart failure in the older population. J Geriatr Cardiol.
11:329–337. 2014.
|
|
2
|
Johansen H, Strauss B, Arnold JM, Moe G
and Liu P: On the rise: The current and projected future burden of
congestive heart failure hospitalization in Canada. Can J Cardiol.
19:430–435. 2003.PubMed/NCBI
|
|
3
|
Kishi T: Heart failure as an autonomic
nervous system dysfunction. J Cardiol. 59:117–122. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Triposkiadis F, Karayannis G, Giamouzis G,
Skoularigis J, Louridas G and Butler J: The sympathetic nervous
system in heart failure physiology, pathophysiology and clinical
implications. J Am Coll Cardiol. 54:1747–1762. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Guyenet PG: The sympathetic control of
blood pressure. Nat Rev Neurosci. 7:335–346. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Potts JT, Paton JF, Mitchell JH, Garry MG,
Kline G, Anguelov PT and Lee SM: Contraction-sensitive skeletal
muscle afferents inhibit arterial baroreceptor signalling in the
nucleus of the solitary tract: Role of intrinsic GABA interneurons.
Neuroscience. 119:201–214. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schreihofer AM and Guyenet PG: The
baroreflex and beyond: Control of sympathetic vasomotor tone by
GABAergic neurons in the ventrolateral medulla. Clin Exp Pharmacol
Physiol. 29:514–521. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Affleck VS, Coote JH and Pyner S: The
projection and synaptic organisation of NTS afferent connections
with presympathetic neurons, GABA and nNOS neurons in the
paraventricular nucleus of the hypothalamus. Neuroscience.
219:48–61. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Braga VA, Medeiros IA, Ribeiro TP,
França-Silva MS, Botelho-Ono MS and Guimarães DD:
Angiotensin-II-induced reactive oxygen species along the
SFO-PVN-RVLM pathway: Implications in neurogenic hypertension. Braz
J Med Biol Res. 44:871–876. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kumagai H, Oshima N, Matsuura T, Iigaya K,
Imai M, Onimaru H, Sakata K, Osaka M, Onami T, Takimoto C, et al:
Importance of rostral ventrolateral medulla neurons in determining
efferent sympathetic nerve activity and blood pressure. Hypertens
Res. 35:132–141. 2012. View Article : Google Scholar :
|
|
11
|
Tagawa T and Dampney RA: AT(1) receptors
mediate excitatory inputs to rostral ventrolateral medulla pressor
neurons from hypothalamus. Hypertension. 34:1301–1307. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shafton AD, Ryan A and Badoer E: Neurons
in the hypothalamic paraventricular nucleus send collaterals to the
spinal cord and to the rostral ventrolateral medulla in the rat.
Brain Res. 801:239–243. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nunn N, Womack M, Dart C and
Barrett-Jolley R: Function and pharmacology of spinally-projecting
sympathetic pre-autonomic neurones in the paraventricular nucleus
of the hypothalamus. Curr Neuropharmacol. 9:262–277. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sun SY, Wang W, Zucker IH and Schultz HD:
Enhanced peripheral chemoreflex function in conscious rabbits with
pacing-induced heart failure. J Appl Physiol (1985). 86:1264–1272.
1999.
|
|
15
|
Reid IA: Interactions between ANG II,
sympathetic nervous system and baroreceptor reflexes in regulation
of blood pressure. Am J Physiol. 262:E763–E778. 1992.PubMed/NCBI
|
|
16
|
Liu JL, Murakami H, Sanderford M, Bishop
VS and Zucker IH: ANG II and baroreflex function in rabbits with
CHF and lesions of the area postrema. Am J Physiol. 277:H342–H350.
1999.PubMed/NCBI
|
|
17
|
Llewellyn TL, Sharma NM, Zheng H and Patel
KP: Effects of exercise training on SFO-mediated sympathoexcitation
during chronic heart failure. Am J Physiol Heart Circ Physiol.
306:H121–H131. 2014. View Article : Google Scholar :
|
|
18
|
Parsons KK and Coffman TM: The
reninangiotensin system: It's all in your head. J Clin Invest.
117:873–876. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lavoie JL, Cassell MD, Gross KW and
Sigmund CD: Adjacent expression of renin and angiotensinogen in the
rostral ventro-lateral medulla using a dual-reporter transgenic
model. Hypertension. 43:1116–1119. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lavoie JL, Cassell MD, Gross KW and
Sigmund CD: Localization of renin expressing cells in the brain, by
use of a REN-eGFP transgenic model. Physiol Genomics. 16:240–246.
2004. View Article : Google Scholar
|
|
21
|
Veerasingham SJ and Raizada MK: Brain
renin-angiotensin system dysfunction in hypertension: Recent
advances and perspectives. Br J Pharmacol. 139:191–202. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zheng H, Li YF, Wang W and Patel KP:
Enhanced angiotensin-mediated excitation of renal sympathetic nerve
activity within the paraventricular nucleus of anesthetized rats
with heart failure. Am J Physiol Regul Integr Comp Physiol.
297:R1364–R1374. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gao L, Wang WZ, Wang W and Zucker IH:
Imbalance of angiotensin type 1 receptor and angiotensin II type 2
receptor in the rostral ventrolateral medulla: Potential mechanism
for sympathetic overactivity in heart failure. Hypertension.
52:708–714. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang WZ, Gao L, Wang HJ, Zucker IH and
Wang W: Interaction between cardiac sympathetic afferent reflex and
chemoreflex is mediated by the NTS AT1 receptors in heart failure.
Am J Physiol Heart Circ Physiol. 295:H1216–H1226. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tan J, Wang H and Leenen FH: Increases in
brain and cardiac AT1 receptor and ACE densities after myocardial
infarct in rats. Am J Physiol Heart Circ Physiol. 286:H1665–H1671.
2004. View Article : Google Scholar
|
|
26
|
Wei SG, Yu Y, Zhang ZH, Weiss RM and
Felder RB: Mitogen-activated protein kinases mediate upregulation
of hypothalamic angiotensin II type 1 receptors in heart failure
rats. Hypertension. 52:679–686. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Isegawa K, Hirooka Y, Katsuki M, Kishi T
and Sunagawa K: Angiotensin II type 1 receptor expression in
astrocytes is upregulated leading to increased mortality in mice
with myocardial infarction-induced heart failure. Am J Physiol
Heart Circ Physiol. 307:H1448–H1455. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ramchandra R, Hood SG, Watson AM, Allen AM
and May CN: Central angiotensin type 1 receptor blockade decreases
cardiac but not renal sympathetic nerve activity in heart failure.
Hypertension. 59:634–641. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li Z, Iwai M, Wu L, Shiuchi T, Jinno T,
Cui TX and Horiuchi M: Role of AT2 receptor in the brain in
regulation of blood pressure and water intake. Am J Physiol Heart
Circ Physiol. 284:H116–H121. 2003. View Article : Google Scholar
|
|
30
|
Gao J, Zhang H, Le KD, Chao J and Gao L:
Activation of central angiotensin type 2 receptors suppresses
norepinephrine excretion and blood pressure in conscious rats. Am J
Hypertens. 24:724–730. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gao J, Zucker IH and Gao L: Activation of
central angiotensin type 2 receptors by compound 21 improves
arterial baroreflex sensitivity in rats with heart failure. Am J
Hypertens. 27:1248–1256. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gao L, Wang W, Wang W, Li H, Sumners C and
Zucker IH: Effects of angiotensin type 2 receptor overexpression in
the rostral ventrolateral medulla on blood pressure and urine
excretion in normal rats. Hypertension. 51:521–527. 2008.
View Article : Google Scholar
|
|
33
|
Kang J, Posner P and Sumners C:
Angiotensin II type 2 receptor stimulation of neuronal K+ currents
involves an inhibitory GTP binding protein. Am J Physiol.
267:C1389–C1397. 1994.PubMed/NCBI
|
|
34
|
Qi J, Zhang DM, Suo YP, Song XA, Yu XJ,
Elks C, Lin YX, Xu YY, Zang WJ, Zhu Z and Kang YM:
Renin-angiotensin system modulates neurotransmitters in the
paraventricular nucleus and contributes to angiotensin II-induced
hypertensive response. Cardiovasc Toxicol. 13:48–54. 2013.
View Article : Google Scholar
|
|
35
|
Chen Q and Pan HL: Signaling mechanisms of
angiotensin II-induced attenuation of GABAergic input to
hypothalamic presympathetic neurons. J Neurophysiol. 97:3279–3287.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hu L, Zhu DN, Yu Z, Wang JQ, Sun ZJ and
Yao T: Expression of angiotensin II type 1 (AT(1)) receptor in the
rostral ventrolateral medulla in rats. J Appl Physiol (1985).
92:2153–2161. 2002. View Article : Google Scholar
|
|
37
|
Paton JF, Deuchars J, Ahmad Z, Wong LF,
Murphy D and Kasparov S: Adenoviral vector demonstrates that
angiotensin II-induced depression of the cardiac baroreflex is
mediated by endothelial nitric oxide synthase in the nucleus
tractus solitarii of the rat. J Physiol. 531:445–458. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Paton JF, Boscan P, Murphy D and Kasparov
S: Unravelling mechanisms of action of angiotensin II on
cardiorespiratory function using in vivo gene transfer. Acta
Physiol Scand. 173:127–137. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chan SH, Hsu KS, Huang CC, Wang LL, Ou CC
and Chan JY: NADPH oxidase-derived superoxide anion mediates
angiotensin II-induced pressor effect via activation of p38
mitogen-activated protein kinase in the rostral ventrolateral
medulla. Circ Res. 97:772–780. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gao L, Li Y, Schultz HD, Wang WZ, Wang W,
Finch M, Smith LM and Zucker IH: Downregulated Kv4.3 expression in
the RVLM as a potential mechanism for sympathoexcitation in rats
with chronic heart failure. Am J Physiol Heart Circ Physiol.
298:H945–H955. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kang YM, Ma Y, Zheng JP, Elks C, Sriramula
S, Yang ZM and Francis J: Brain nuclear factor-kappa B activation
contributes to neurohumoral excitation in angiotensin II-induced
hypertension. Cardiovasc Res. 82:503–512. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gao L, Wang W, Li YL, Schultz HD, Liu D,
Cornish KG and Zucker IH: Superoxide mediates sympathoexcitation in
heart failure: Roles of angiotensin II and NAD(P)H oxidase. Circ
Res. 95:937–944. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang G, Anrather J, Glass MJ, Tarsitano
MJ, Zhou P, Frys KA, Pickel VM and Ladecola C: Nox2, Ca2+ and
protein kinase C play a role in angiotensin II-induced free radical
production in nucleus tractus solitarius. Hypertension. 48:482–489.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu D, Gao L, Roy SK, Cornish KG and
Zucker IH: Role of oxidant stress on AT1 receptor expression in
neurons of rabbits with heart failure and in cultured neurons. Circ
Res. 103:186–193. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Nishihara M, Hirooka Y, Matsukawa R, Kishi
T and Sunagawa K: Oxidative stress in the rostral ventrolateral
medulla modulates excitatory and inhibitory inputs in spontaneously
hypertensive rats. J Hypertens. 30:97–106. 2012. View Article : Google Scholar
|
|
46
|
Gao L, Wang W, Liu DM and Zucker IH:
Exercise training normalizes sympathetic outflow by central
antioxidant mechanisms in rabbits with pacing-induced chronic heart
failure. Circulation. 115:3095–3102. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Li Y, Zhang W and Stern JE: Nitric oxide
inhibits the firing activity of hypothalamic paraventricular
neurons that innervate the medulla oblongata: Role of GABA.
Neuroscience. 118:585–601. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Krukoff TL and Khalili P: Stress-induced
activation of nitric oxide-producing neurons in the rat brain. J
Comp Neurol. 377:509–519. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lin LH, Taktakishvili O and Talman WT:
Identification and localization of cell types that express
endothelial and neuronal nitric oxide synthase in the rat nucleus
tractus solitarii. Brain Res. 1171:42–51. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chan SH, Wang LL and Chan JY: Differential
engagements of glutamate and GABA receptors in cardiovascular
actions of endogenous nNOS or iNOS at rostral ventrolateral medulla
of rats. Br J Pharmacol. 138:584–593. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Patel KP, Li YF and Hirooka Y: Role of
nitric oxide in central sympathetic outflow. Exp Biol Med
(Maywood). 226:814–824. 2001.
|
|
52
|
Wang Y, Patel KP, Cornish KG, Channon KM
and Zucker IH: nNOS gene transfer to RVLM improves baroreflex
function in rats with chronic heart failure. Am J Physiol Heart
Circ Physiol. 285:H1660–H1667. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sakai K, Hirooka Y, Shigematsu H, Kishi T,
Ito K, Shimokawa H, Takeshita A and Sunagawa K: Overexpression of
eNOS in brain stem reduces enhanced sympathetic drive in mice with
myocardial infarction. Am J Physiol Heart Circ Physiol.
289:H2159–H2166. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wang Y, Liu XF, Cornish KG, Zucker IH and
Patel KP: Effects of nNOS antisense in the paraventricular nucleus
on blood pressure and heart rate in rats with heart failure. Am J
Physiol Heart Circ Physiol. 288:H205–H213. 2005. View Article : Google Scholar
|
|
55
|
Zhang K, Li YF and Patel KP: Blunted
nitric oxide-mediated inhibition of renal nerve discharge within
PVN of rats with heart failure. Am J Physiol Heart Circ Physiol.
281:H995–H1004. 2001.PubMed/NCBI
|
|
56
|
Hirooka Y, Shigematsu H, Kishi T, Kimura
Y, Ueta Y and Takeshita A: Reduced nitric oxide synthase in the
brainstem contributes to enhanced sympathetic drive in rats with
heart failure. J Cardiovasc Pharmacol. 42(Suppl 1): S111–S115.
2003. View Article : Google Scholar
|
|
57
|
Zucker IH, Schultz HD, Li YF, Wang Y, Wang
W and Patel KP: The origin of sympathetic outflow in heart failure:
The roles of angiotensin II and nitric oxide. Prog Biophys Mol
Biol. 84:217–232. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Jaffrey SR, Snowman AM, Eliasson MJ, Cohen
NA and Snyder SH: CAPON: A protein associated with neuronal nitric
oxide synthase that regulates its interactions with PSD95. Neuron.
20:115–124. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Sharma NM, Zheng H, Mehta PP, Li YF and
Patel KP: Decreased nNOS in the PVN leads to increased
sympathoexcitation in chronic heart failure: Role for CAPON and Ang
II. Cardiovasc Res. 92:348–357. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Sharma NM, Llewellyn TL, Zheng H and Patel
KP: Angiotensin II-mediated posttranslational modification of nNOS
in the PVN of rats with CHF: Role for PIN. Am J Physiol Heart Circ
Physiol. 305:H843–H855. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Horn T, Smith PM, McLaughlin BE, Bauce L,
Marks GS, Pittman QJ and Ferguson AV: Nitric oxide actions in
paravenstricular nucleus: Cardiovascular and neurochemical
implications. Am J Physiol. 266:R306–R313. 1994.PubMed/NCBI
|
|
62
|
Zhang K and Patel KP: Effect of nitric
oxide within the para-ventricular nucleus on renal sympathetic
nerve discharge: Role of GABA. Am J Physiol. 275:R728–R734.
1998.
|
|
63
|
Li YF, Mayhan WG and Patel KP:
NMDA-mediated increase in renal sympathetic nerve discharge within
the PVN: Role of nitric oxide. Am J Physiol Heart Circ Physiol.
281:H2328–H2336. 2001.
|
|
64
|
Zheng H, Liu X, Li Y, Sharma NM and Patel
KP: Gene transfer of neuronal nitric oxide synthase to the
paraventricular nucleus reduces the enhanced glutamatergic tone in
rats with chronic heart failure. Hypertension. 58:966–973. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kishi T, Hirooka Y, Sakai K, Shigematsu H,
Shimokawa H and Takeshita A: Overexpression of eNOS in the RVLM
causes hypotension and bradycardia via GABA release. Hypertension.
38:896–901. 2001.PubMed/NCBI
|
|
66
|
Martins-Pinge MC, Garcia MR, Zoccal DB,
Crestani CC and Pinge-Filho P: Differential influence of iNOS and
nNOS inhibitors on rostral ventrolateral medullary mediated
cardiovascular control in conscious rats. Auton Neurosci.
131:65–69. 2007. View Article : Google Scholar
|
|
67
|
Sharma NM, Zheng H, Li YF and Patel KP:
Nitric oxide inhibits the expression of AT1 receptors in neurons.
Am J Physiol Cell Physiol. 302:C1162–C1173. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Guo ZL, Tjen-A-Looi SC, Fu LW and
Longhurst JC: Nitric oxide in rostral ventrolateral medulla
regulates cardiac-sympathetic reflexes: Role of synthase isoforms.
Am J Physiol Heart Circ Physiol. 297:H1478–H1486. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wang S, Paton JF and Kasparov S:
Differential sensitivity of excitatory and inhibitory synaptic
transmission to modulation by nitric oxide in rat nucleus tractus
solitarii. Exp Physiol. 92:371–382. 2007. View Article : Google Scholar
|
|
70
|
Dias AC, Vitela M, Colombari E and Mifflin
SW: Nitric oxide modulation of glutamatergic, baroreflex and
cardiopulmonary transmission in the nucleus of the solitary tract.
Am J Physiol Heart Circ Physiol. 288:H256–H262. 2005. View Article : Google Scholar
|
|
71
|
Ramchandra R, Hood SG and May CN: Central
exogenous nitric oxide decreases cardiac sympathetic drive and
improves baroreflex control of heart rate in ovine heart failure.
Am J Physiol Regul Integr Comp Physiol. 307:R271–R280. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Rauchhaus M, Doehner W, Francis DP, Davos
C, Kemp M, Liebenthal C, Niebauer J, Hooper J, Volk HD, Coats AJ
and Anker SD: Plasma cytokine parameters and mortality in patients
with chronic heart failure. Circulation. 102:3060–3067. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Utsuyama M and Hirokawa K: Differential
expression of various cytokine receports in the brain after
stimulation with LPS in young and old mice. Exp Gerontol.
37:411–420. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Wei SG, Zhang ZH, Beltz TG, Yu Y, Johnson
AK and Felder RB: Subfornical organ mediates sympathetic and
hemo-dynamic responses to blood-borne proinflammatory cytokines.
Hypertension. 62:118–125. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Felder RB, Yu Y, Zhang ZH and Wei SG:
Pharmacological treatment for heart failure: A view from the brain.
Clin Pharmacol Ther. 86:216–220. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Yu Y, Zhang ZH, Wei SG, Serrats J, Weiss
RM and Felder RB: Brain perivascular macrophages and the
sympathetic response to inflammation in rats after myocardial
infarction. Hypertension. 55:652–659. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zhang ZH, Yu Y, Wei SG and Felder RB:
Centrally administered lipopolysaccharide elicits sympathetic
excitation via NAD(P)H oxidase-dependent mitogen-activated protein
kinase signaling. J Hypertens. 28:806–816. 2010. View Article : Google Scholar :
|
|
78
|
Francis J, Zhang ZH, Weiss RM and Felder
RB: Neural regulation of the proinflammatory cytokine response to
acute myocardial infarction. Am J Physiol Heart Circ Physiol.
287:H791–H797. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Kang YM, Zhang ZH, Xue B, Weiss RM and
Felder RB: Inhibition of brain proinflammatory cytokine synthesis
reduces hypothalamic excitation in rats with ischemia-induced heart
failure. Am J Physiol Heart Circ Physiol. 295:H227–H236. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Kang YM, Ma Y, Elks C, Zheng JP, Yang ZM
and Francis J: Cross-talk between cytokines and renin-angiotensin
in hypo-thalamic paraventricular nucleus in heart failure: Role of
nuclear factor-kappaB. Cardiovasc Res. 79:671–678. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Guggilam A, Cardinale JP, Mariappan N,
Sriramula S, Haque M and Francis J: Central TNF inhibition results
in attenuated neurohumoral excitation in heart failure: A role for
superoxide and nitric oxide. Basic Res Cardiol. 106:273–286. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Allen RG and Tresini M: Oxidative stress
and gene regulation. Free Radic Biol Med. 28:463–499. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Akira S and Takeda K: Toll-like receptor
signalling. Nat Rev Immunol. 4:499–511. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Ogawa K, Hirooka Y, Kishi T and Sunagawa
K: Brain AT1 receptor activates the sympathetic nervous system
through toll-like receptor 4 in mice with heart failure. J
Cardiovasc Pharmacol. 58:543–549. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Kang YM, He RL, Yang LM, Qin DN, Guggilam
A, Elks C, Yan N, Guo Z and Francis J: Brain tumour necrosis
factor-alpha modulates neurotransmitters in hypothalamic
paraventricular nucleus in heart failure. Cardiovasc Res.
83:737–746. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Wei SG, Zhang ZH, Yu Y and Felder RB:
Central SDF-1/CXCL12 expression and its cardiovascular and
sympathetic effects: The role of angiotensin II, TNF-α and MAPK
signaling. Am J Physiol Heart Circ Physiol. 307:H1643–H1654. 2014.
View Article : Google Scholar : PubMed/NCBI
|