Introduction
Polyploidization contributes to genetic instability
in cancer by causing the unequal distribution of chromosomes in
cell divisions (1–3). That linkage supports a role of
mechanisms that augment the number of tumor genomes per cell, e.g.,
cell fusion and failed mitosis/absence of cell division following
genome replication in increasing the degree of aneuploidy (2). Intriguingly, upon increasing the
content of DNA by heterotypic cell fusion, cancer cells undergo
large-scale changes in gene expression that induce progression
(4,5). These findings implicate cell fusion
as a mechanism of cancer cell progression towards highly malignant
phenotypes, thus suggesting that certain fusogenic mechanistic
underpinnings of cells may be a feature of carcinogenic
transformation. Molecular studies have elucidated sequential
cell-cell fusion steps in fusogenic cells that contribute to
mammalian development, tissue repair and homeostasis (6–8),
and have provided experimental support for the possibility of an
association between cell fusion and cancer cell transformation
(6). However, the molecular
determinants of cell-cell fusion in cancer remain unknown.
In a previous study, in a set of human cancer cell
lines, we found a correlation between the proportion of cells with
genome duplication and the proportion of cells invading through
proteolytic degradation (9).
Thus, the contribution of invasion to polyploidization indicated
that tumor-genome duplication was mediated by intracellular
invasion, which led us to hypothesize that adhesion signaling may
induce polyploidization in cancer cells. In this study, employing
expression vectors of a reporter array for transcription factors of
signaling networks implicated in cancer, we pinpointed pathways of
signal transduction in glioblastoma cells to investigate whether
adhesion signaling determines the frequency of
polyploidization.
Materials and methods
Reagents
Integrin-β1 polyclonal antibody was obtained from
Cell Signaling Technology, Inc. (Danvers, MA, USA); antibody for
hyaluronan receptor (clone G44–26) was from BD Biosciences (San
Jose, CA, USA); antibody for neuronal cell adhesion molecule
(Nr-CAM) (clone A27), marimastat and matrix metalloproteinase (MMP)
inhibitor II were purchased from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA); non-specific antibodies were obtained from
Rockland Immunochemicals, Inc. (phycoerythrin-conjugated)
(Gilbertsville, PA, USA), and Santa Cruz Biotechnology, Inc.
Tissue culture
The cell cultures were maintained at 37°C in a
humidified atmosphere containing 5% CO2. U87MG cells
(American Type Culture Collection, Manassas, VA, USA) were
maintained in minimum essential medium (Mediatech, Herndon, VA,
USA) supplemented with 10% fetal bovine serum (Atlanta Biologicals,
Inc., Flowery Branch, GA, USA), 2 mM L-glutamine (HyClone, Logan,
UT, USA), 50 U/ml penicillin and 50 μg/ml streptomycin (both from
Lonza, Rockland, ME, USA). Neural progenitor cells from newborn
C57BL/6 mice (Harlan Laboratories, Indianapolis, IN, USA) were
obtained, after euthanasia following a protocol approved by the
Animal Care and Use Committee of our Institution, through
incubation of sections of the forebrain in neurobasal medium
supplemented with B27 (both from Invitrogen Life Technologies,
Carlsbad, CA, USA), 20 ng/ml epidermal growth factor, 10 ng/ml
basic fibroblast growth factor (both from R&D Systems,
Minneapolis, MN, USA), 2 mM L-glutamine, 50 U/ml penicillin and 50
μg/ml streptomycin. Fluorescent marker-expressing cells, obtained
by clonal expansion (U87MG cells) or fluorescence-activated cell
sorting (neural progenitor cells) after transduction with
retroviral vectors as previously described (10), were co-cultured in neurobasal
medium. Tissue culture trypsinization was carried out with Hank’s
balanced salt solution containing 0.05% trypsin (Mediatech), and
terminated by the addition of serum-supplemented culture medium
(U87MG cells) or by dilution (neural progenitor cells).
Transduction with luciferase reporter
vector
Stably transduced reporter cells were obtained by
cell culture incubation in medium containing lentiviral particles
with luciferase expression vector (Cignal Finder Cancer Reporter
Array; SABiosciences, Frederick, MD, USA), and the selection of the
population of transduced cells through the addition of puromycin
(0.5 μg/ml; Sigma-Aldrich, St. Louis, MO, USA).
Luciferase assay
Luciferase expression by cells transduced with
reporter vector, containing either non-inducible TATA-box promoter
or TATA-box promoter joined to one of 10 different
transcriptional-response elements, was quantified in 96-well plate
adherent cultures. For this purpose, a 1:1 mixture of ONE-Glo™
Luciferase reagent (Promega Corp., Madison, WI, USA) and cell
culture medium supplemented with 2% fetal bovine serum was added to
the cells (50 μl/cell culture) and, 10 min later, the luminescence
of the resulting cell lysate was measured in a Turner Biosystems
20/20 luminometer (Turner Biosystems, Inc., Sunnyvale, CA,
USA).
Determination of cell density-dependent
transcriptional activation
In a series consisting of 5 individual cultures of
lentiviral vector-transduced cells set up with cell densities
decreasing, from 59,200 cells/cm2, by a 2-fold serial
dilution factor, luciferase reporter gene expression was determined
after 48 h from cell seeding, at which time cell confluency was
expected to be 64, 32, 16, 8 and 4% (control) (based on 34 h
doubling time and 250,000 cells/cm2 in a confluent
culture). The fold change in cell expression of reporter was
determined by dividing the value of luminescence by the average
value of the corresponding control cultures (4% cell confluency)
and by the corresponding dilution. A statistical threshold for
transcriptional activation was established from all the average
ratios obtained as the median ± SD. Average ratios above the
threshold were considered indicative of promoter activation.
Protein determination
Total protein amount per cell culture was estimated
using the BCA protein assay kit (Pierce Biotechnology, Inc.,
Rockford, IL, USA), after rinsing each culture with
phosphate-buffered saline (PBS) and lysing the cells with cold
lysis buffer (pH 7.4) containing 20 mM Tris, 0.5% Triton X-100, 150
mM NaCl and protease inhibitor cocktail (1:100; EMD Millipore,
Billerica, MA, USA).
Plate coating
Untreated polystyrene plates were coated with the
adhesion protein, retronectin (Takara, Otsu, Japan). Retronectin
was added at different dilutions in PBS to non-tissue
culture-treated plates (BD Biosciences), removing the solutions
after 2 h of coating at room temperature. The coated surfaces were
rinsed with PBS before seeding the cells. The retronectin coating
concentrations were determined as indicated by the
manufacturer.
Cell DNA content distribution
analysis
Cell DNA content was quantified by measuring the
emission intensity of fluorescent dye-stained DNA in a flow
cytometer (Guava EasyCyte™ Plus System; EMD Millipore). For this
purpose, the cell culture was trypsinized and the cell suspension
passed through a cell strainer (70 μm) (BD Biosciences). After
blocking trypsinization, 2×104 cells were centrifuged
and resuspended in 0.5 ml of PBS. Using a fine tip pipette, the
suspension was drawn up and expelled repeatedly. Subsequently, upon
the addition of a 10-fold larger volume of buffer, the cells were
centrifuged and resuspended in 200 μl of PBS. The suspension was
added drop by drop to 10 ml of ice-cold 70% ethanol for cell
fixation. The fixed cells were centrifuged at high speed (450 × g),
resuspended in PBS containing 1% bovine serum albumin,
re-centrifuged and resuspended in 200 μl of Guava cell cycle
reagent (EMD Millipore). The distribution of propidium
iodide-stained cells was determined using Guava CytoSoft 5.3
software (Guava Technologies, Inc., Hayward, CA, USA).
Estimation of the fraction of binucleated
cells
The fraction of binucleated cells over the total
cells was quantified in 3-day cultures on a retronectin-coated
surface (seeding density, 15,000 cells/cm2). Each
culture was trypsinized and re-seeded in a regular culture well
and, soon after attachment, the cells were fixed with 4%
p-formaldehyde and stained with 4′,6-diamidino-2-phenylindole
(1:1,000; Sigma-Aldrich). Binucleated cells were counted in
randomly selected areas using an Olympus CKX41 fluorescence
microscope (Olympus, Center Valley, PA, USA).
Quantitative assessment of receptor
expression
The cell surface expression of hyaluronan receptor
was quantified by flow cytometric analysis. To this aim,
2×104 cells/sample were resuspended in PBS containing
0.1% bovine serum albumin and, then, half of the cells were
incubated for 30 min at 4°C with phycoerythrin-conjugated antibody
for hyaluronan receptor and the other half with
phycoerythrin-conjugated non-specific antibody (antibody
concentration, 0.5 μg/ml). Following incubation with antibody, the
cells were washed twice and resuspended in 200 μl of PBS for the
analysis of fluorescence intensity in a Guava flow cytometer.
Statistical analysis
The Student’s t-test was used to determine p-values.
p<0.05 was considered to indicate a statistically significant
result.
Results
Intercellular integrin binding activates
both ERK and Notch target transcription in U87MG glioblastoma
cells
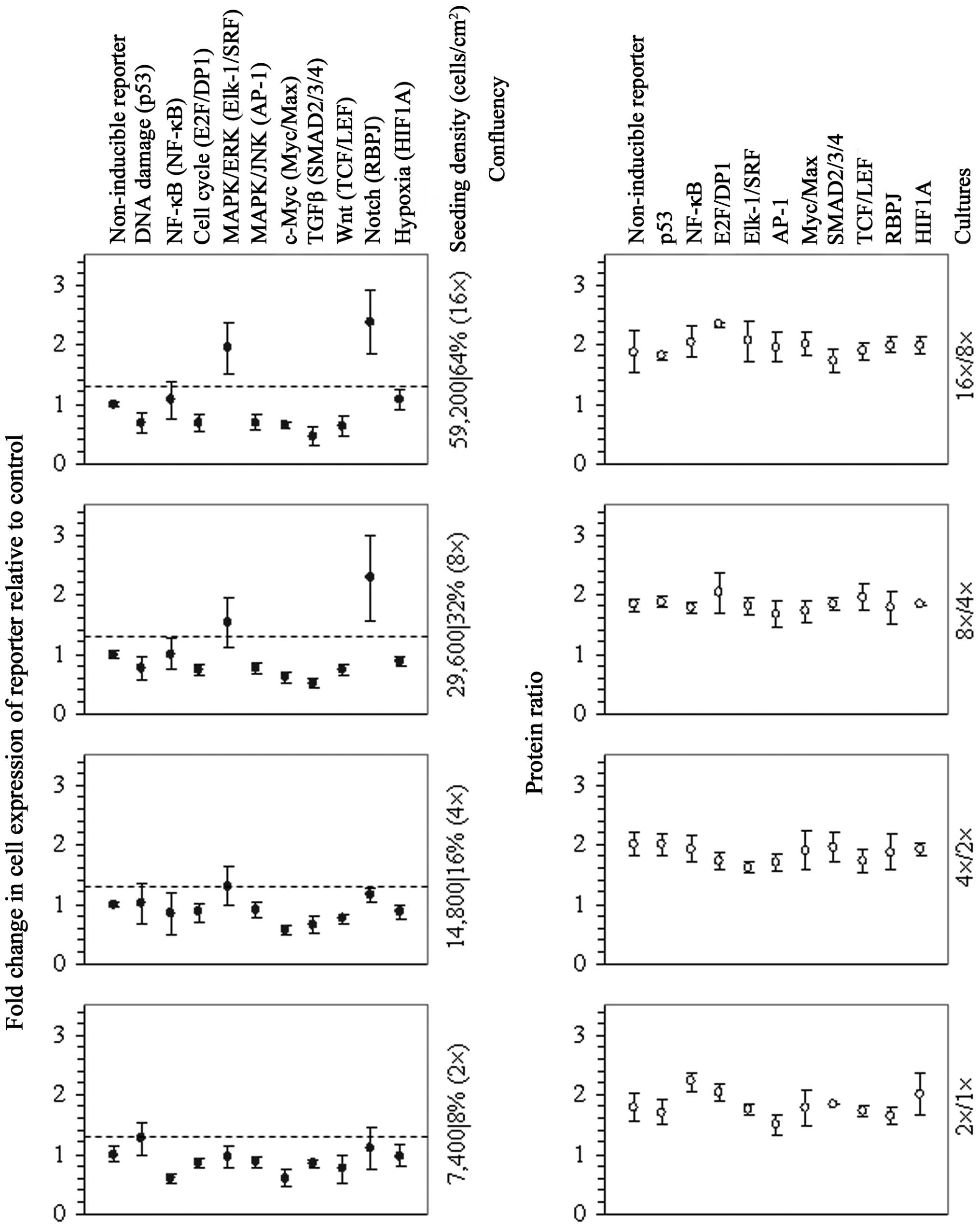
The assessment of transcription of vectors of a
pathway reporter array in 2-fold serial dilution cultures revealed
both the activation of Elk-1/SRF- and RBPJ-driven transcription in
the cell cultures at 32% confluency and further enhanced
transcription in the cell cultures at 64% confluency (Fig. 1), indicating that intercellular
adhesion elicited signal transduction through either extracellular
signal-regulated kinase (ERK), Notch or both pathways. Given that
cell adhesion-induced ERK activation is determined by integrin
binding to protein ligands (11),
we subsequently assessed cell density-dependent transcriptional
activation in cultures containing integrin-β1 antibody. Antibody
targeting of integrin-β1 hindered the induction of both ERK and
Notch target transcription (Table
I), suggesting that integrin-mediated signal transduction
activated ERK and that, concomitantly, there was either a crosstalk
between the ERK and Notch signaling pathways or an interaction
between the integrin and Notch receptors (12). Since Nr-CAM is a binding partner
of integrins in cell-cell adhesion (13), we also determined cell
density-dependent transcriptional activation in cultures containing
a specific antibody for that adhesion receptor. This corroborated
that signaling decreased (Table
I) upon the antibody targeting of a ligand of integrins at the
cell surface. Conversely, there was signaling activation in the
cultures containing non-specific antibody or blocking antibody for
hyaluronan receptor (Table I), a
mediator of cell-cell adhesion that has been related to cell fusion
(7,14), which indicated that cell
confluence and signal transduction were not impaired by the
side-effects of the antibody. In summary, these results revealed
that intercellular integrin binding elicited adhesion signaling in
U87MG cells that activated both ERK and Notch target
transcription.
 | Table IDecreased signaling in U87MG
glioblastoma cell cultures upon antibody targeting of integrin-β1
or Nr-CAM. |
Table I
Decreased signaling in U87MG
glioblastoma cell cultures upon antibody targeting of integrin-β1
or Nr-CAM.
| Fold increase in
transcription factor-regulated expression relative to the
controla |
|---|
|
|
|---|
| Antibody | Elk-1/SRF | RBPJ |
|---|
| Non-specific | 1.8±0.4 | 2.0±0.2 |
| Integrin-β1 |
1.0±0.1b |
0.8±0.2c |
| Nr-CAM |
0.9±0.2b |
1.1±0.2c |
| Hyaluronan
receptor | 1.8±0.2 | 1.6±0.3 |
Enhanced frequency of protease-driven
polyploidization upon activation of both ERK and Notch target
transcription
We assessed the level of both ERK and Notch target
transcription following integrin-based cell adhesion to surfaces
coated with retronectin, a recombinant adhesion protein with
integrin binding sites of fibronectin, which is a pericellular
matrix component robustly expressed by U87MG cells (15). Cell attachment to coating at a
concentration of 0.5 μg/cm2 involved transcriptional
activation (Table II), providing
the induction of both ERK and Notch target transcription. We then
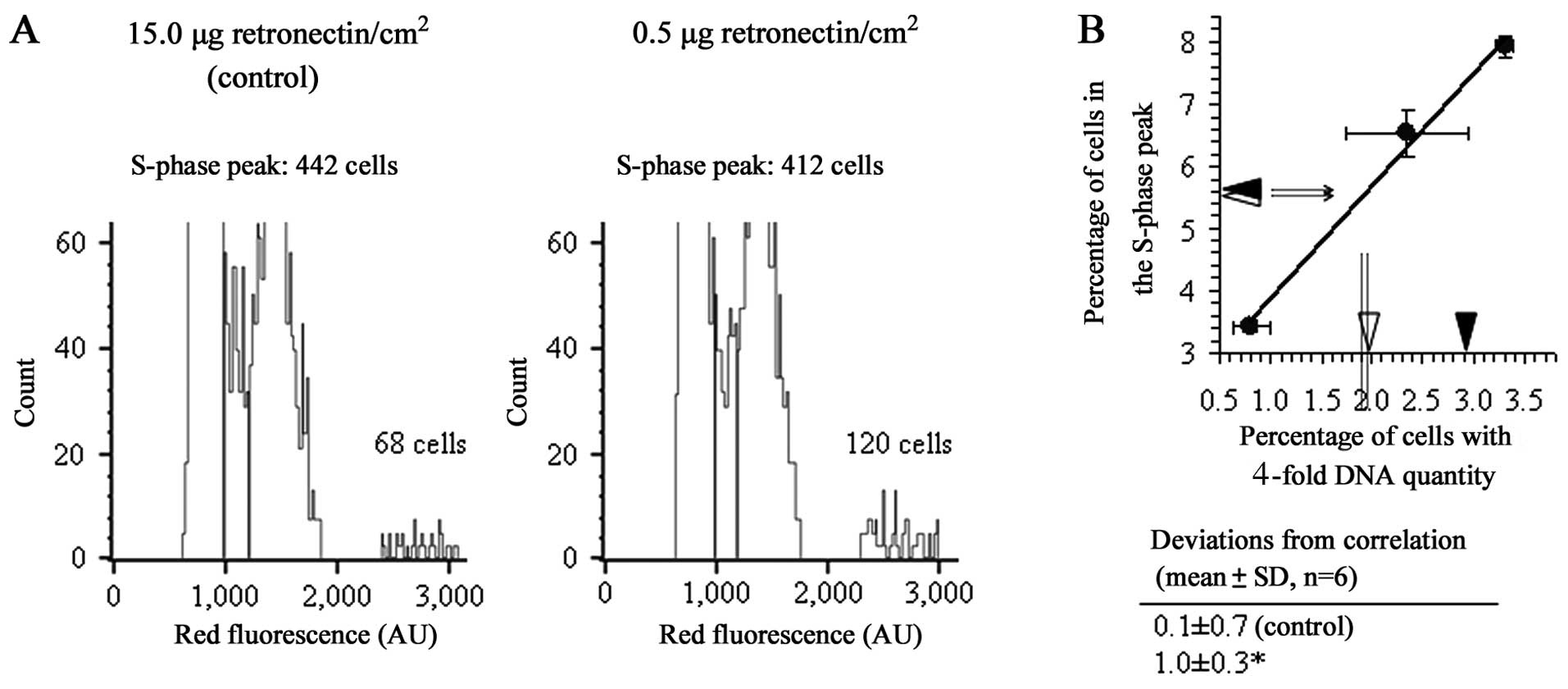
determined the rate of proliferation and the percentage of cells
with a 4-fold increase in the amount of cellular DNA, corresponding
to the G2/M peak of the cells with duplicated tumor genome
(9), of cultures on coating that
promoted integrin adhesion signaling. The fold increase in the
number of cells in the 24–48 h interval from cell seeding, 2.8±0.9,
was almost equal to that of the control cultures (15 μg
retronectin/cm2), 2.9±0.7 (means ± SD of 3 different
experiments). The relative number of cells with duplicated tumor
genome, by contrast, increased significantly (Fig. 2). In agreement with this result,
the cultures contained a markedly enhanced proportion of
binucleated cells (Table III).
This linked the transcriptional activation induced by integrin
adhesion to an increased rate of cell polyploidization. Hence, we
quantified the cells with a duplicated tumor genome in regular
cultures containing antibody that prevented signaling activation.
The cultures containing antibody for hyaluronan receptor, which
prevented the polyploidization of breast carcinoma and melanoma
cells, showed an unaltered frequency of polyploidization (Table IV). By contrast, the cultures
containing integrin-β1 antibody showed a reduced frequency
(Table V). These findings
indicated that the induction of both ERK and Notch target
transcription resulted in tumor-genome duplication.
| Table IIInduction of both ERK and Notch target
transcription in U87MG cells by integrin-mediated adhesion to
retronectin coating. |
Table II
Induction of both ERK and Notch target
transcription in U87MG cells by integrin-mediated adhesion to
retronectin coating.
| Retronectin
concentration (μg/cm2) | Fold change in
transcription factor-regulated expression relative to the
controla |
|---|
|
|---|
| Elk-1/SRF | RBPJ |
|---|
| 8.0 | 0.9±0.1 | 1.1±0.2 |
| 4.5 | 1.0±0.2 | 0.9±0.3 |
| 3.0 | 1.0±0.1 | 1.1±0.3 |
| 1.5 | 0.9±0.1 | 1.0±0.1 |
| 0.5 | 1.4±0.1b | 1.7±0.2b |
| Table IIIU87MG cell cultures on coating that
promoted integrin adhesion signaling show enhanced frequency of
polynucleation. |
Table III
U87MG cell cultures on coating that
promoted integrin adhesion signaling show enhanced frequency of
polynucleation.
| 15.0 μg
Retronectin/cm2 | 0.5 μg
Retronectin/cm2 |
|---|
|
|
|---|
| Binucleated/total
cells | % | Binucleated/total
cells | % |
|---|
| 22/388 | 6 | 38/361 | 11 |
| 11/236 | 5 | 31/315 | 10 |
| 31/458 | 7 | 32/283 | 11 |
| 35/390 | 9 | 31/292 | 11 |
| 13/278 | 5 | 56/334 | 17 |
| 22/334 | 7 | 22/271 | 8 |
| Means ± SD | 7±2 | | 11±3a |
| Table IVDespite expressing hyaluronan
receptor, as determined by analysis using phycoerythrin-conjugated
antibody, U87MG glioblastoma cell cultures containing specific
antibody show unchanged frequency of polyploidizationa. |
Table IV
Despite expressing hyaluronan
receptor, as determined by analysis using phycoerythrin-conjugated
antibody, U87MG glioblastoma cell cultures containing specific
antibody show unchanged frequency of polyploidizationa.
| Fluorescence
intensity (a.u.) | Cells with 4-fold
increase in DNA quantity (percentage of control)b |
|---|
|
|
|
|---|
| Antibody | U87MG | MDA | FEMX | U87MG | MDA | FEMX |
|---|
| Non-specific | 2±1 | 2±1 | 1±0 | 95±23 | 100±19 | 99±16 |
| Hyaluronan
receptor | 831±268 | 827±261 | 393±105 | 106±9 | 58±7c | 51±22c |
| Table VReduced proportion of cells with a
4-fold increase in DNA quantity in U87MG cell cultures containing
antibody for integrin-β1. |
Table V
Reduced proportion of cells with a
4-fold increase in DNA quantity in U87MG cell cultures containing
antibody for integrin-β1.
| Antibody | Cells with 4-fold
increase in DNA quantity (percentage of control)a |
|---|
| Non-specific | 109±19 |
| Integrin-β1 | 77±13b |
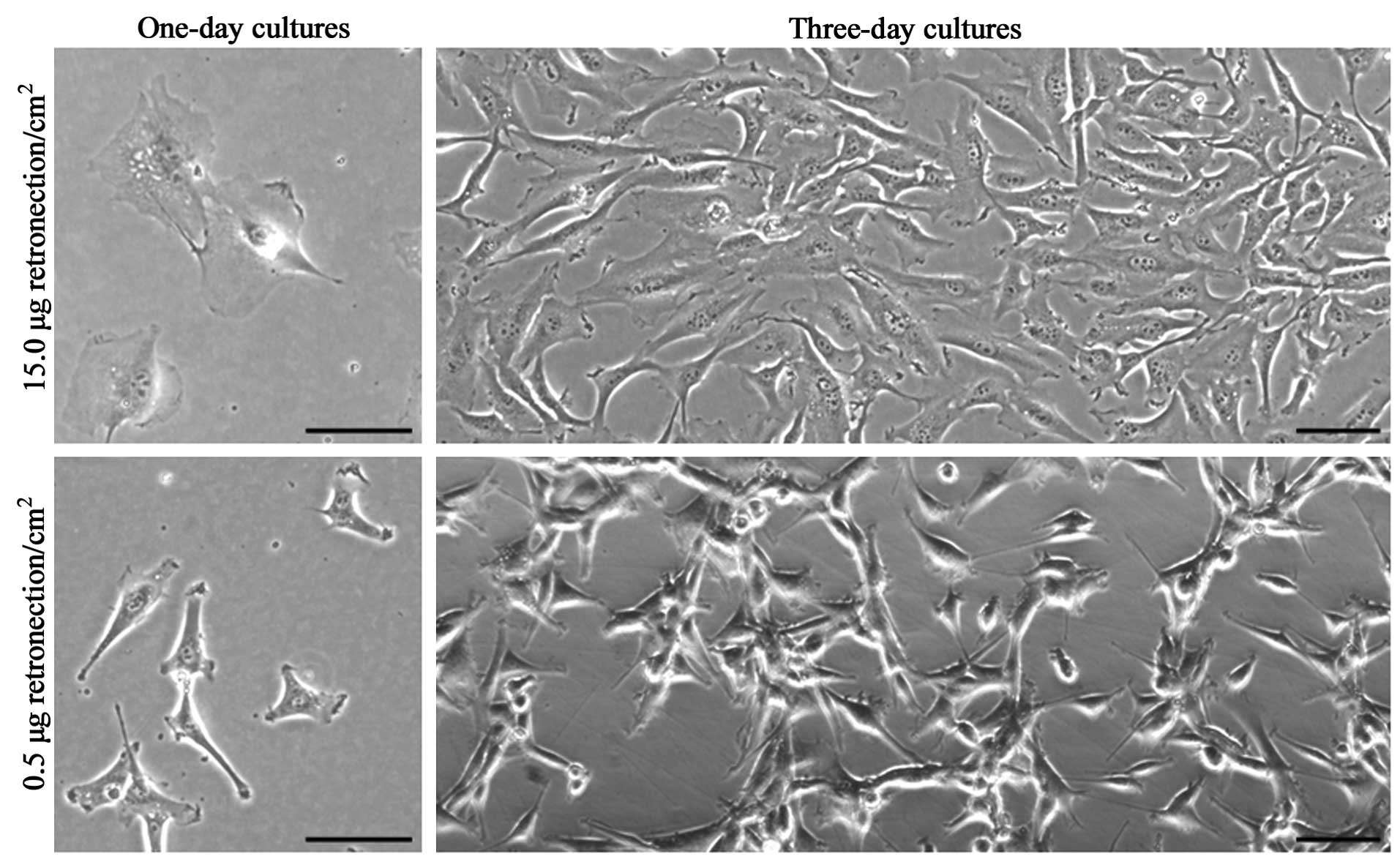
Of note, the U87MG cells on coating that promoted
integrin adhesion signaling extended thin protruding adhesive
structures resembling matrix-degrading invadopodia (Fig. 3), which suggested that the
induction of genome duplication was mediated by intracellular
invasion. Hence, we quantified the cells with a duplicated tumor
genome in the regular cell cultures containing specific inhibitors
of cancer cell invasion-associated MMPs. Cultures containing MMP
inhibitors had markedly low percentages of polyploid cells
(Table VI). Since these findings
indicated that adhesion-stimulated genome duplication was mediated
by protease-driven tumor invasion, we wished to investigate whether
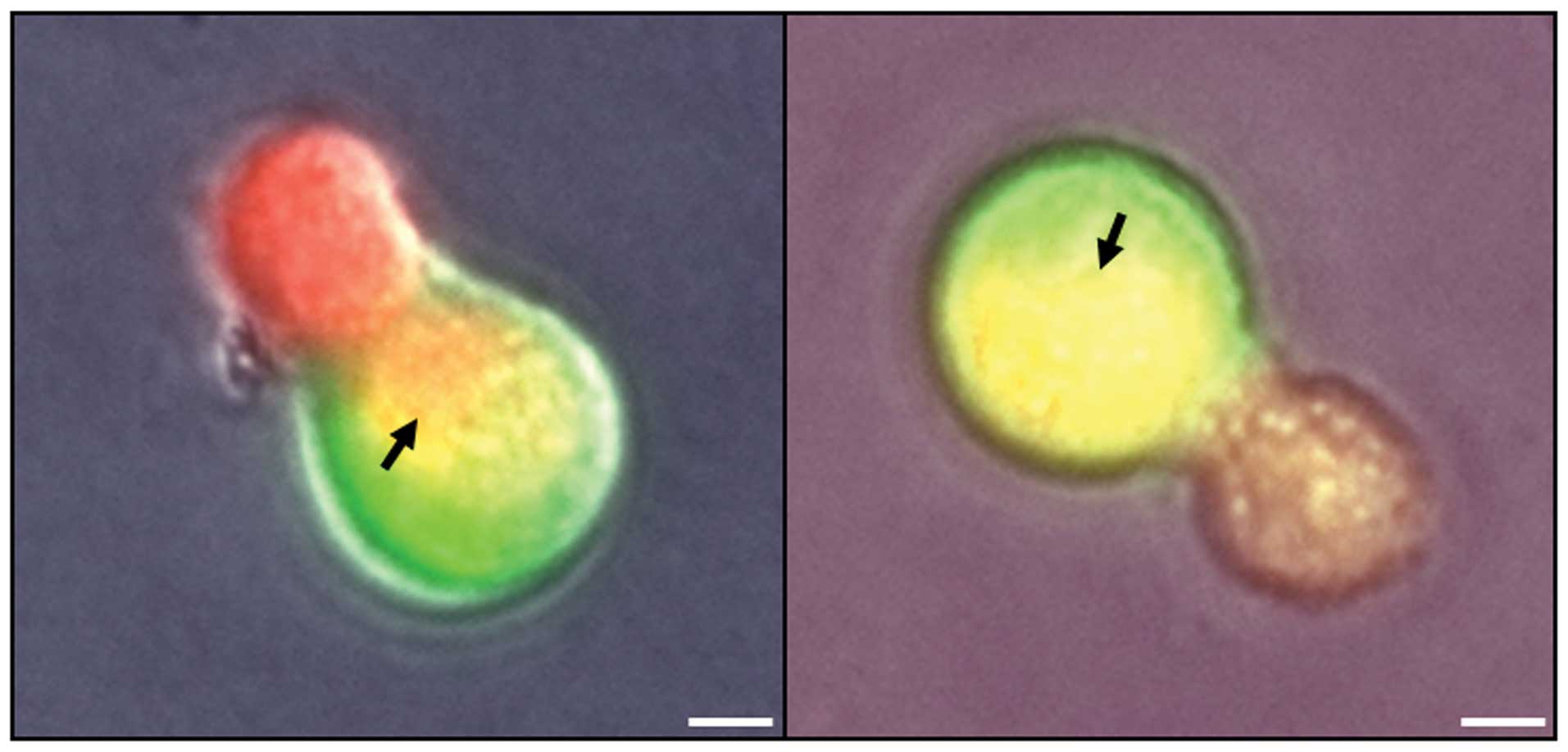
intracellular invasion by U87MG cells resulted in the merging of
cell contents. In co-cultures in non-coated dishes of U87MG cells
expressing red fluorescent protein and non-tumor neural cells
expressing green fluorescent protein, intracellular fluorescence
was observed that revealed that the cytoplasmic content of tumor
cells was released through fusion pores. Representative images are
presented in Fig. 4. These
observations revealed that intracellular invasion by U87MG cells
resulted in the merging of cell contents, further suggesting that
tumor invasion mediated the increases in DNA content.
| Table VIReduced proportion of cells with a
4-fold increase in DNA quantity in U87MG cell cultures containing
MMP inhibitor. |
Table VI
Reduced proportion of cells with a
4-fold increase in DNA quantity in U87MG cell cultures containing
MMP inhibitor.
| Compound | Cells with 4-fold
increase in DNA quantity (percentage of control)a |
|---|
| Chemical
solvent | 100±20 |
| MMP inhibitor
II | 59±19b |
| Marimastat | 62±6b |
Discussion
In the present study, we found that signal
transduction promoted the polyploidization of U87MG glioblastoma
cells. Specifically, the activation of both ERK and Notch target
transcription induced by integrin adhesion resulted in a
significantly enhanced number of hypotetraploid cells. This
indicated that adhesion-stimulated polyploidization was associated
with the activation of signaling networks that regulate the
expression of proteolytic enzymes that elicit invasion, since Notch
signaling regulates the expression of adamalysin proteins (16), which are reportedly profusogenic
metalloproteases (6,8) and ERK signaling involves the
transcriptional control of MMP expression (17–20). Accordingly, inhibitors for the
specific blockage of several types of cancer cell
invasion-associated MMPs markedly reduced the frequency of
tumor-genome duplication. Thus, consistent with the observation
that intracellular invasion by U87MG cells resulted in the merging
of cell contents, both the finding that transcriptional activation
and podosome-like adhesive structures appeared together, and the
inhibitory effect of MMP blockage revealed that the induction of
tumor-genome duplication was mediated by protease-driven invasion.
Our results therefore indicated that the activation of cell
invasion-regulating signaling networks determined the appearance of
polyploid cells.
Both the antibody targeting of Nr-CAM, a ligand of
integrins in cell-cell adhesion that is overexpressed in human
brain tumors (21) and
integrin-β1, a molecule that also mediates integrin receptor
adhesion to matrix proteins (11), hindered signaling activation in
U87MG cell cultures. This suggested that both intercellular
integrin binding to proteins of the plasma membrane and
pericellular matrix elicited the transduction of signals that
induced polyploidization. We previously found that the antibody
targeting of hyaluronan receptor significantly reduced the
proportion of polyploid cells in both breast carcinoma and melanoma
cell cultures (9), thus
supporting a role of that adhesion receptor in cancer
invasion-mediated cell fusion. In this study, by contrast, the
antibody did not hinder the polyploidization of glioblastoma cells.
As the expression of hyaluronan receptor on the cell surface was
not exceedingly high, this result seemed unrelated to the level of
receptor saturation reached by the antibody. Instead, the lack of
inhibitory effect suggested that intercellular interactions of
hyaluronan receptor did not result in polyploidization.
In conclusion, the findings of the present study
demonstrate the association of the activation of invasion signaling
upon cell adhesion with the appearance of tumor-genome duplication,
supporting cell invasiveness as a cause of ploidy turnover in tumor
cell populations.
Abbreviations:
|
ERK
|
extracellular signal-regulated
kinase
|
|
MMP
|
matrix metalloproteinase
|
|
Nr-CAM
|
neuronal cell adhesion molecule
|
|
PBS
|
phosphate-buffered saline
|
References
|
1
|
Sluder G and Nordberg JJ: The good, the
bad and the ugly: the practical consequences of centrosome
amplification. Curr Opin Cell Biol. 16:49–54. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Davoli T and de Lange T: The causes and
consequences of polyploidy in normal development and cancer. Annu
Rev Cell Dev Biol. 27:585–610. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vitale I, Galluzzi L, Senovilla L, Criollo
A, Jemaà M, Castedo M and Kroemer G: Illicit survival of cancer
cells during polyploidization and depolyploidization. Cell Death
Differ. 18:1403–1413. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chakraborty AK, Sodi S, Rachkovsky M,
Kolesnikova N, Platt JT, Bolognia JL and Pawelek JM: A spontaneous
murine melanoma lung metastasis comprised of host x tumor hybrids.
Cancer Res. 60:2512–2519. 2000.PubMed/NCBI
|
|
5
|
Lagarde AE, Donaghue TP, Dennis JW and
Kerbel RS: Genotypic and phenotypic evolution of a murine tumor
during its progression in vivo toward metastasis. J Natl Cancer
Inst. 71:183–191. 1983.PubMed/NCBI
|
|
6
|
Ogle BM, Cascalho M and Platt JL:
Biological implications of cell fusion. Nat Rev Mol Cell Biol.
6:567–575. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen EH, Grote E, Mohler W and Vignery A:
Cell-cell fusion. FEBS Lett. 581:2181–2193. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou X and Platt JL: Molecular and
cellular mechanisms of mammalian cell fusion. Adv Exp Med Biol.
713:33–64. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mercapide J, Anzanello F, Rappa G and
Lorico A: Relationship between tumor cell invasiveness and
polyploidization. PLoS One. 7:e533642012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lorico A, Mercapide J, Solodushko V,
Alexeyev M, Fodstad O and Rappa G: Primary neural stem/progenitor
cells expressing endostatin or cytochrome P450 for gene therapy of
glioblastoma. Cancer Gene Ther. 15:605–615. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yee KL, Weaver VM and Hammer DA:
Integrin-mediated signalling through the MAP-kinase pathway. IET
Syst Biol. 2:8–15. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Campos LS, Decker L, Taylor V and Skarnes
W: Notch, epidermal growth factor receptor, and beta1-integrin
pathways are coordinated in neural stem cells. J Biol Chem.
281:5300–5309. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Conacci-Sorrell M, Kaplan A, Raveh S,
Gavert N, Sakurai T and Ben-Ze’ev A: The shed ectodomain of Nr-CAM
stimulates cell proliferation and motility, and confers cell
transformation. Cancer Res. 65:11605–11612. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sterling H, Saginario C and Vignery A:
CD44 occupancy prevents macrophage multinucleation. J Cell Biol.
143:837–847. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Enam SA, Rosenblum ML and Edvardsen K:
Role of extracellular matrix in tumor invasion: migration of glioma
cells along fibronectin-positive mesenchymal cell processes.
Neurosurgery. 42:599–607. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Díaz B, Yuen A, Iizuka S, Higashiyama S
and Courtneidge SA: Notch increases the shedding of HB-EGF by
ADAM12 to potentiate invadopodia formation in hypoxia. J Cell Biol.
201:279–292. 2013.PubMed/NCBI
|
|
17
|
Anand M, van Meter TE and Fillmore HL:
Epidermal growth factor induces matrix metalloproteinase-1 (MMP-1)
expression and invasion in glioma cell lines via the MAPK pathway.
J Neurooncol. 104:679–687. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kunapuli P, Kasyapa CS, Hawthorn L and
Cowell JK: LGI1, a putative tumor metastasis suppressor gene,
controls in vitro invasiveness and expression of matrix
metalloproteinases in glioma cells through the ERK1/2 pathway. J
Biol Chem. 279:23151–23157. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lakka SS, Jasti SL, Gondi C, Boyd D,
Chandrasekar N, Dinh DH, Olivero WC, Gujrati M and Rao JS:
Downregulation of MMP-9 in ERK-mutated stable transfectants
inhibits glioma invasion in vitro. Oncogene. 21:5601–5608. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lin YM, Jan HJ, Lee CC, Tao HY, Shih YL,
Wei HW and Lee HM: Dexamethasone reduced invasiveness of human
malignant glioblastoma cells through a MAPK phosphatase-1 (MKP-1)
dependent mechanism. Eur J Pharmacol. 593:1–9. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sehgal A, Boynton AL, Young RF, Vermeulen
SS, Yonemura KS, Kohler EP, Aldape HC, Simrell CR and Murphy GP:
Cell adhesion molecule Nr-CAM is over-expressed in human brain
tumors. Int J Cancer. 76:451–458. 1998. View Article : Google Scholar : PubMed/NCBI
|