Introduction
microRNAs (miRNAs) are non-coding, small,
single-stranded, endogenous RNAs composed of 20–25 nucleotides.
miRNAs regulate gene expression at the post-transcriptional level
through sequence-specific interactions with 3′-untranslated regions
(UTRs) in mRNAs and also via translation inhibition or degradation
of mRNAs (1–4). Several studies have suggested that
aberrantly expressed miRNAs may act as oncogenes or tumor
suppressor genes in lung cancers (5–7).
Accumulating data have revealed that miRNAs regulate a variety of
biological processes, including cell proliferation, apoptosis,
development and differentiation (8,9).
Thus, deregulation of miRNA expression may play a causal role in
carcinogenesis, growth and metastasis (10,11).
Numerous genes have target sites for interaction with miRNAs and an
individual miRNA is capable of modulating the expression of
multiple genes (12); thus, the
miRNA regulatory network is highly complex.
Our previous study using microarray technology,
revealed that miR-99b is downregulated in patients with lung cancer
(7), and that fibroblast growth
factor receptor 3 (FGFR3) is a predicted target of miR-99b in three
public algorithms (Pictar, TargetScan and miRanda). FGFR3 is a
member of the transmembrane receptor kinase for the FGF family of
ligands, which play key roles in the regulation of cell
proliferation, differentiation and tumorigenesis (13–15).
In the present study, we determined the differential expression of
miR-99b and FGFR3 in lung cancer and normal lung tissues. We
hypothesized that miR-99b plays a role in cell growth in lung
cancer by directly targeting FGFR3. To test this hypothesis, we
assessed cell proliferation and performed a colony formation
assay.
Materials and methods
Cell lines
H1299, H522, HCC95 and HCC1438 lung cancer cells
were maintained in RPMI-1640 medium (Gibco-BRL, Glasgow, UK) with
10% fetal bovine serum (FBS) and antibiotics (100 units/ml
penicillin and 100 g/ml streptomycin).
Tissues
Tumor and corresponding normal lung tissue specimens
were obtained from 27 patients with non-small cell lung cancer
(NSCLC). A total of 27 patients with NSCLC (9 squamous cell
carcinomas, 17 adenocarcinomas and 1 large cell carcinoma), who
underwent curative resection at the Konyang University Hospital
(Daejeon, Korea), were analyzed. None of the patients had received
chemotherapy or radiotherapy prior to surgery. Informed consent was
obtained from each patient prior to surgery. This study was
approved by the Bioethics Committee of Konyang University Hospital.
All of the tumor and macroscopically normal lung tissue samples
were obtained at the time of surgery, and were rapidly frozen in
liquid nitrogen and stored at −80°C until analysis. Tissue samples
were histologically confirmed by hematoxylin and eosin
staining.
microRNA precursor transfection
Cells were plated in 6-well plates at a density of
1.5×105 cells/well. The following day, cells were
transfected with 50 nM pre-miR™ miRNA precursor (has-miR-99b;
Ambion, Austin, TX, USA) and pre-miR™miRNA precursor-negative
control #1 (negative control #1; Ambion) with Oligofectamine
(Invitrogen, Carlsbad, CA, USA) based on the manufacturer’s
instructions.
Quantitative real-time polymerase chain
reaction (qRT-PCR) assays
Total RNA was isolated with TRIzol reagent
(Gibco-BRL) according to the manufacturer’s instructions. The first
strand of complementary DNA (cDNA) was synthesized using the
oligo(dT) primer system (SuperScript III First-strand Synthesis
System; Invitrogen). Aliquots of the reaction mixture were used for
the quantitative PCR (qPCR) amplification with the IQ5 system
(Bio-Rad Laboratories, Hercules, CA, USA) using iQ SYBR-Green
Supermix (Bio-Rad). PCR was run for 40 cycles of denaturation at
95°C for 15 sec, annealing at 55°C for 15 sec, and elongation at
72°C for 15 sec. Gene expression was quantified by the comparative
CT method, with normalizing CT values to the housekeeping gene,
β-actin. Following amplification, melting curve analysis was
performed to ensure the specificity of the products.
TaqMan microRNA expression assay
qRT-PCR analysis for miRNAs was performed in
duplicate with a TaqMan MicroRNA assay kit (Applied Biosystems,
Foster City, CA, USA) according to the manufacturer’s instructions,
and RNU6B was used for normalization.
Luciferase assay
We determined whether or not miR99b modulates FGFR3
expression using a luciferase assay. The dual luciferase vector,
psiCHECK2, was purchased from Promega (Madison, WI, USA). The 1463
bp fragment was synthesized by PCR using cDNA of 293T cells. The
forward primer with the XhoI restriction site
(5′-GGGCTCGAGGGCC ACTGGTCCCCAACAATGTG-3′) and the reverse primer
with the NotI restriction site (5′-GGGCGGCCGCCCAGTAA
CAGTACAGAACGAACCAAC-3′) were used to amplify the FGFR 3′UTR. The
PCR products were cloned into the XhoI/NotI 3′UTR
site of the psiCHECK2 plasmid (Promega). The correct sequence of
all the clones was verified by DNA sequencing. 293T cells were
seeded in 12-well plates in Dulbecco’s modified Eagle’s medium
(DMEM) medium, supplemented with 10% heat-inactivated FBS. The
cells were transfected with psiCHECK2-FGFR UTR constructs
containing 3′-UTR of FGFR, in the presence or absence of miR99b
(Ambion) using Effectene (Qiagen, Hilden, Germany). The cells were
collected 48 h following transfection, and the cell lysates were
prepared according to the Promega instruction manual. The Renilla
luciferase activity was measured using a Lumat LB953 luminometer
(EG&G Berthhold, Bad Wildbad, Germany), and the results were
normalized using the activity of luciferase. All experiments were
performed in triplicate.
Westren blot analyses
Cells were lysed in Pro-Prep protein extraction
solution (iNtRON Biotechnology, Gyeonnggi-do, Korea) 48 h following
transfection. An equal amount of proteins was resolved by 8% sodium
dodecyl sulfate polyacrylamide gel electrophoresis gels. The
primary antibodies used for the analysis were mouse anti-FGFR3
(1:1000; BD Biosciences, San Jose, CA, USA) and mouse anti-β-actin
antibodies (1:2000; Santa Cruz Biotechnology, Inc., Santa Cruz, CA,
USA).
MTS assay
Cells were seeded in 96-well plates at a density
which enabled transfection per well in triplicate. Proliferation
indices were measured 24, 47 and 72 h later using the CellTiter96
Aqueous One Solution Cell Proliferation Assay (MTS assay; Promega).
All experiments were performed in triplicate.
Colony formation assay
We used the Cell Transformation Detection kit
(Millipore, Bedford, MA, USA) to evaluate the ability to form
colonies on soft agar. Briefly, 1 ml underlayers consisting of 0.8%
agar medium were prepared in 6-well plates. miR-99b and negative
control miRNA-transfected cells were trypsinized, centrifuged,
resuspended in 0.4% agar medium (equal volumes of 0.8% Noble agar
and culture medium), and plated onto the top agar at 2,500 cells
per well. The cells were kept wet by adding a small amount of
RPMI-1640 with 10% FBS and incubated for 3 weeks at 37°C. Fresh
culture medium was replaced every 3 days. Colonies were visualized
using cell staining solution (Millipore) and counted under a
microscope.
Statistical analyses
Statistical differences in the expression of miRNAs
were analyzed by the Student’s t-test. Statistical analysis was
performed using SPSS 12.0 computer software (SPSS Inc.; Chicago,
IL, USA). A value of p<0.05 was considered to be
significant.
Result
Quantitative analysis of miR-99b and
FGFR3 expression in human lung cancer
qRT-PCR was applied to detect the miRNA and FGFR3
expression in 27 pairs of lung cancer tissue samples and matched
normal lung tissue samples. miR-99b was significantly downregulated
in lung cancer tissues (p=0.013), while FGFR3 was upregulated in
lung cancer tissues (p= 0.015; Fig.
1).
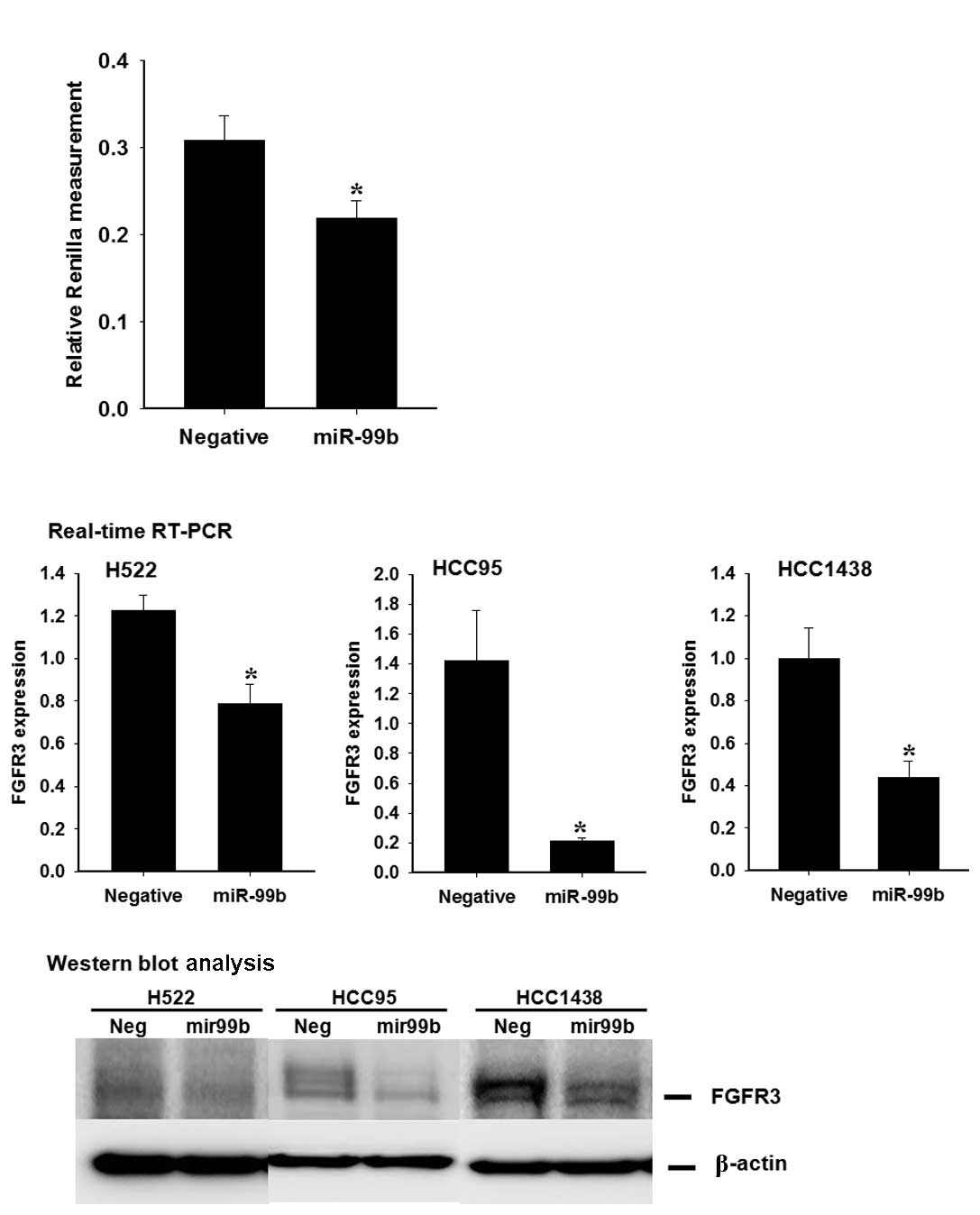
FGFR3 transcription is repressed by
binding of miR-99b to the 3′-UTR
To verify that miR-99b is capable of regulating the
FGFR3 gene directly, we generated a Rellina luciferase
reporter plasmid, cloned downstream to a segment of the FGFR
3′UTR containing the putative miR-99b binding sequence. The
constructs were then co-transfected into 293T cells with
psiCHECK2-mir-99b or psiCHECK2, and Rellina luciferase activity was
measured 48 h later. As shown in Fig.
2, significantly lower luciferase activity was generated by the
miR-99b precursor as compared with negative miRNA (p<0.0001).
This result indicates that miR-99b suppresses FGFR3 transcription
by direct binding to the 3′-UTR.
miR-99b regulates FGFR3 expression at the
mRNA and protein levels
miRNAs are capable of suppressing the expression of
target genes through translational repression or degradation of
target transcripts. To assess whether or not miR-99b has a
functional role in the downregulation of endogenous FGFR3
expression, HCC95, HCC1428 and H522 cells were transfected with the
miR-99b precursor, the expression of FGFR3 was then measured by
qRT-PCR and Western blot analysis. When miR-99b was overexpressed,
FGFR3 expression was diminished compared with the control group
(Fig. 3).
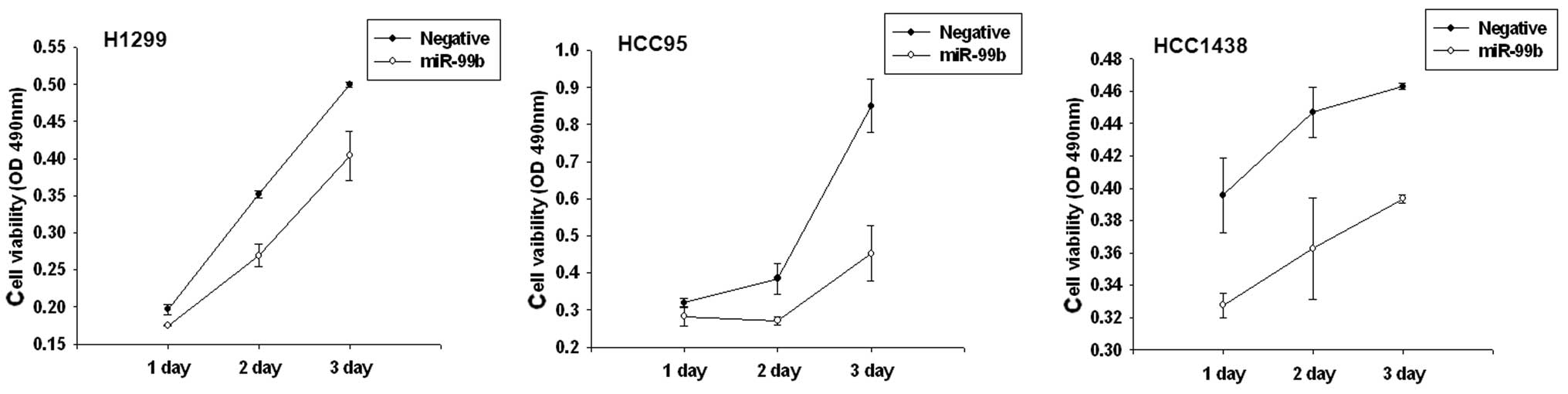
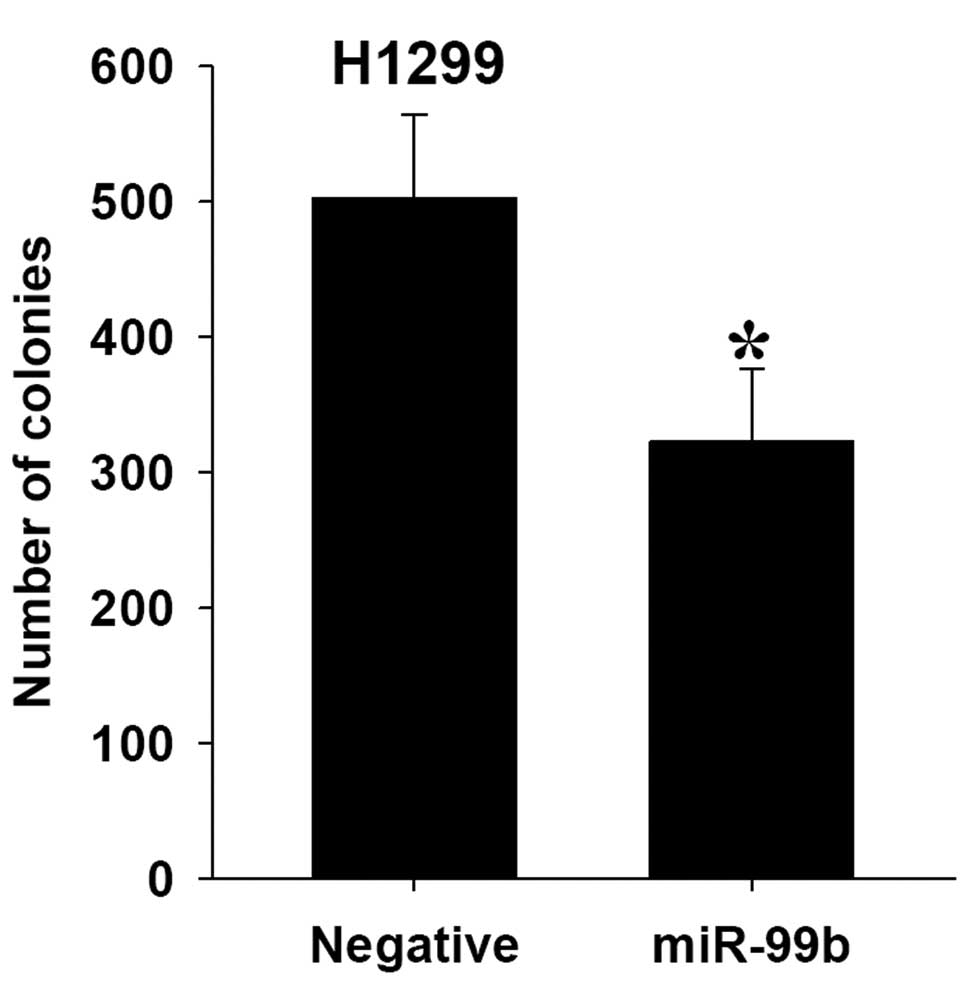
Alteration of miR-99b affects lung cell
growth and colony formation in vitro
To determine whether overexpression of miR99b has an
effect on cell growth in lung cancer, we performed a proliferation
assay in lung cancer cell lines. Based on the results of the MTS
assay and growth curves, we demonstrated that cells which were
transiently transfected with the miR-99b precursor, had a
significant growth inhibition at varying degrees in H1299, HCC95
and HCC1438 cell lines. Furthermore, miR-99b affected
anchorage-independent growth. Transfection with the miR-99b
precursor markedly decreased the plating efficiency of H1299 cells
(64.1%; Fig. 4).
Discussion
Self-sufficiency in growth signals is the hallmark
of cancer and is frequently driven by receptor tyrosine kinase
(RTK)-dependent growth factor pathways (16). These growth factor pathways are an
attractive target for lung cancer treatment, including
small-molecule tyrosine kinase inhibitors (TKIs) and monoclonal
antibodies. Several miRNAs have been reported to regulate
RTK-dependent growth factor pathways. miR-7 directly regulates the
epidermal growth factor receptor (EGFR) and Raf1, and indirectly
regulates EGFR signaling at multiple levels, including PI3K, ERK
and AKT. miR-7 inhibits cell cycle progression and reduces cell
growth and viability (17).
miR-145 targets the EGFR and inhibits cell proliferation in
transfected lung adenocarcinoma cells (18). miR-221 and 222 induce TNF-related
apoptosis-inducing ligand (TRAIL) resistance through activation of
the AKT pathway by targeting phosphatase and tensin homolog (PTEN)
tumor suppressors (19). In
addition, miR-1 has been reported to target ectopic c-Met and
induce apoptosis in response to anticancer drugs (20). These findings suggest a potential
therapeutic application of miRNAs against lung cancers through the
inhibition of RTK-dependent growth factor pathways.
FGFR3 belongs to a family of structurally related
tyrosine kinase receptors (FGFR1–4), which are involved in numerous
aspects of various biological processes, including proliferation,
differentiation, migration and apoptosis (21,22).
These receptors consist of an extracellular domain that includes a
signal peptide followed by three immunoglobulin (Ig)-like domains,
an acidic box, a transmembrane domain and an intracellular tyrosine
kinase domain (15). FGFR3 is
activated by FGF ligand binding to extracellular Ig-like domains II
and III. Subsequently, trans-autophosphorylation at tyrosine
residues in the cytoplasmic domain is required for the stimulation
of the intrinsic catalytic activity and activation of downstream
signaling pathways. FGFR3 has been demonstrated to be involved in
the RAS/RAF/MEK/MAPK pathway through the activation of p90
ribosomal S6 kinase (23).
There are certain somatic gain-of-function mutations
that cause the activation of FGFR3 in the absence of a ligand in
bladder tumors. The activating point mutations are frequently
observed in non-invasive, low-grade and low-stage bladder tumors.
With respect to hematological malignancies, chromosomal
translocation t(4;14) was shown in 15–20% of multiple myeloma
patients (24,25). Notably, a considerable proportion
of bladder tumors with no detectable mutation displayed
overexpression, including numerous muscle-invasive tumors (26). Even though the FGFR3 mutation has
not been reported in lung cancer, FGFR3 is overexpressed in lung
cancer (27,28). This evidence suggests that a loss
of regulatory factors of the FGFR3 may exist in lung cancer. Based
on our results, the expressions of miR-99b and FGFR3 were inversely
correlated in lung cancer and FGFR3 was directly regulated by
miR-99b. These findings suggest that the downregulation of miR-99b
is implicated in lung cancer carcinogenesis through the regulation
of FGFR3.
The FGF-FGFR autocrine pathway is known to be
related to EGFR TKI resistance (29,30).
FGF and FGFR are frequently co-expressed in EGFR
TKI-resistant-NSCLC cell lines. Furthermore, a subgroup of NSCLC
cell lines, which frequently exhibit a more mesenchymal
differentiation have been shown to activate the FGF-FGFR autocrine
pathway (31). Moreover, Thomson
et al (29) reported that
FGFR signaling is activated and functional in
epithelial-mesenchymal transition (EMT)-like transition cells,
induced by TGF-β. EMT is known to be involved in cancer
progression, metastasis and chemoresistance (32). Thus, the negative regulation of the
FGFR may result in a change in cancer phenotype and drug
susceptibility.
In the current study, miR-99b was underexpressed and
FGFR3 was overexpressed in patients with lung cancer. We observed a
substantial FGFR3 suppression by miR-99b at the mRNA and protein
levels in lung cancer. Moreover, FGFR3 was directly regulated by
miR-99b through binding of the 3′-UTR. The overexpression of
miR-99b induced growth arrest. Therefore, miR-99b may act as a
tumor suppressor through its regulation of FGFR3.
Acknowledgements
This study was supported by the
National Research Foundation of Korea Grant, funded by the Korean
Government (NRF-2009-0072810)
References
|
1.
|
V AmbrosThe functions of animal
microRNAsNature431350355200410.1038/nature0287115372042
|
|
2.
|
VN KimJ-W NamGenomics of microRNATrends
Genet22165173200610.1016/j.tig.2006.01.00316446010
|
|
3.
|
AJ SchetterNHH HeegaardCC
HarrisInflammation and cancer: interweaving microRNA, free radical,
cytokine and p53
pathwaysCarcinogenesis313749201010.1093/carcin/bgp27219955394
|
|
4.
|
PB KwakS IwasakiY TomariThe microRNA
pathway and cancerCancer
Sci10123092315201010.1111/j.1349-7006.2010.01683.x20726859
|
|
5.
|
S VoliniaGA CalinC-G LiuA microRNA
expression signature of human solid tumors defines cancer gene
targetsProc Natl Acad Sci
USA10322572261200610.1073/pnas.051056510316461460
|
|
6.
|
N YanaiharaN CaplenE BowmanUnique microRNA
molecular profiles in lung cancer diagnosis and prognosisCancer
Cell9189198200610.1016/j.ccr.2006.01.02516530703
|
|
7.
|
J-W SonY-J KimH-M ChoMicroRNA expression
profiles in Korean non-small cell lungTuberc Respir
Dis67413421200910.4046/trd.2009.67.5.413
|
|
8.
|
AM ChengMW ByromJ SheltonLP FordAntisense
inhibition of human miRNAs and indications for an involvement of
miRNA in cell growth and apoptosisNucleic Acids
Res3312901297200510.1093/nar/gki20015741182
|
|
9.
|
C-Z ChenL LiHF LodishDP BartelMicroRNAs
modulate hematopoietic lineage
differentiationScience3038386200410.1126/science.109190314657504
|
|
10.
|
A Esquela-KerscherP TrangJF WigginsThe
let-7 microRNA reduces tumor growth in mouse models of lung
cancerCell Cycle7759764200810.4161/cc.7.6.583418344688
|
|
11.
|
L MaJ YoungH PrabhalamiR-9, a
MYC/MYCN-activated microRNA, regulates E-cadherin and cancer
metastasisNat Cell Biol12247256201020173740
|
|
12.
|
BP LewisCB BurgeDP BartelConserved seed
pairing, often flanked by adenosines, indicates that thousands of
human genes are microRNA
targetsCell1201520200510.1016/j.cell.2004.12.03515652477
|
|
13.
|
FR LamontDC TomlinsonPA CooperSD ShnyderJD
ChesterMA KnowlesSmall molecule FGF receptor inhibitors block
FGFR-dependent urothelial carcinoma growth in vitro and in vivoBr J
Cancer1047582201110.1038/sj.bjc.660601621119661
|
|
14.
|
DM OrnitzJ XuJS ColvinReceptor specificity
of the fibroblast growth factor familyJ Biol
Chem2711529215297199610.1074/jbc.271.25.152928663044
|
|
15.
|
EM HaugstenA WiedlochaS OlsnesJ
WescheRoles of fibroblast growth factor receptors in
carcinogenesisMol Cancer
Res814391452201010.1158/1541-7786.MCR-10-016821047773
|
|
16.
|
D HanahanRA WeinbergThe hallmarks of
cancerCell1005770200010.1016/S0092-8674(00)81683-9
|
|
17.
|
RJ WebsterKM GilesKJ PricePM ZhangJS
MattickPJ LeedmanRegulation of epidermal growth factor receptor
signaling in human cancer cells by microRNA-7J Biol
Chem28457315741200910.1074/jbc.M80428020019073608
|
|
18.
|
WC ChoAS ChowJS AuMiR-145 inhibits cell
proliferation of human lung adenocarcinoma by targeting EGFR and
NUDT1RNA Biol8125131201110.4161/rna.8.1.14259
|
|
19.
|
M GarofaloG Di LevaG RomanomiR-221&222
regulate TRAIL resistance and enhance tumorigenicity through PTEN
and TIMP3 downregulationCancer Cell164985092009
|
|
20.
|
MW NasserJ DattaG NuovoDown-regulation of
micro-RNA-1 (miR-1) in lung cancerJ Biol
Chem2833339433405200818818206
|
|
21.
|
C BillereyD ChopinM-H
Aubriot-LortonFrequent FGFR3 mutations in papillary non-invasive
bladder (pTa) tumorsAm J
Pathol15819551959200110.1016/S0002-9440(10)64665-211395371
|
|
22.
|
D CappellenC De OliveiraD RicolFrequent
activating mutations of FGFR3 in human bladder and cervix
carcinomasNat Genet231820199910.1038/1261510471491
|
|
23.
|
S KangS DongT-L GuFGFR3 activates RSK2 to
mediate hematopoietic transformation through tyrosine
phosphorylation of RSK2 and activation of the MEK/ERK pathwayCancer
Cell12201214200710.1016/j.ccr.2007.08.00317785202
|
|
24.
|
L ChengS ZhangDD DavidsonMolecular
determinants of tumor recurrence in the urinary bladderFuture
Oncol5843857200910.2217/fon.09.5019663734
|
|
25.
|
M ChesiE NardiniLA BrentsFrequent
translocation t(4;14)(p16.3;q32.3) in multiple myeloma is
associated with increased expression and activating mutations of
fibroblast growth factor receptor 3Nat
Genet16260264199710.1038/ng0797-2609207791
|
|
26.
|
DC TomlinsonO BaldoP HarndenMA
KnowlesFGFR3 protein expression and its relationship to mutation
status and prognostic variables in bladder cancerJ
Pathol2139198200710.1002/path.220717668422
|
|
27.
|
M KarouiH Hofmann-RadvanyiU ZimmermannNo
evidence of somatic FGFR3 mutation in various types of
carcinomaOncogene2050595061200110.1038/sj.onc.120465111526491
|
|
28.
|
M WoenckhausJ MerkR StoehrPrognostic value
of FHIT, CTNNB1, and MUC1 expression in non-small cell lung
cancerHum
Pathol39126136200810.1016/j.humpath.2007.05.02717949785
|
|
29.
|
S ThomsonF PettiI Sujka-KwokD EpsteinJ
HaleyKinase switching in mesenchymal-like non-small cell lung
cancer lines contributes to EGFR inhibitor resistance through
pathway redundancyClin Exp
Metastasis25843854200810.1007/s10585-008-9200-418696232
|
|
30.
|
L MarekKE WareA FritzscheFibroblast growth
factor (FGF) and FGF receptor-mediated autocrine signaling in
non-small-cell lung cancer cellsMol
Pharmacol75196207200910.1124/mol.108.04954418849352
|
|
31.
|
SA KonoME MarshallKE WareLE HeasleyThe
fibroblast growth factor receptor signaling pathway as a mediator
of intrinsic resistance to EGFR-specific tyrosine kinase inhibitors
in non-small cell lung cancerDrug Resist
Updat1295102200910.1016/j.drup.2009.05.00119501013
|
|
32.
|
M IwatsukiK MimoriT
YokoboriEpithelial-mesenchymal transition in cancer development and
its clinical significanceCancer
Sci101293299201010.1111/j.1349-7006.2009.01419.x19961486
|