Introduction
During the past several years, extensive effort has
been invested in the field of microRNA (miRNA) polymorphisms and
the risk of various types of human cancers. miRNAs, originally
discovered in Caenorhabditis elegans in 1993 (1) and first reported in 2001 (2), are small, evolutionarily conserved,
single-strand non-coding RNA molecules ∼22 nucleotides in length
(3,4). It is predicted that miRNAs account
for 1–5% of the human genome and regulate at least 30% of
protein-coding genes and play a crucial role in cancer (5–7). To
date, more than one thousand human miRNAs have been identified.
Facilitated by continuing technological advances, to date, most
registered miRNAs have been widely studied and the results show
that both loss and gain of specific miRNA function contribute to
cancer development, shedding more light on cancer prevention,
diagnosis, progression and outcome.
hsa-miR-196a2, discovered by Lagos-Quintana et
al (8), was initially reported
to be a prognostic biomarker for non-small cell lung cancer by Hu
et al (9). Since then,
emerging molecular epidemiological studies have reported the
association between the hsa-miR-196a2 polymorphism and
susceptibility to diverse types of human cancer (10–34).
Although the precise processes controlling miRNA genetic variants
in cancer susceptibility are largely unknown, the importance of
miRNA SNPs has been implicated in many cancers. Common
single-nucleotide polymorphisms (SNPs), such as hsa-miR-196a2
rs11614913 which is located in the pre-miRNA, may affect the
expression and function of mature miRNAs, resulting in diverse
functional consequences, thus opening up a new door through which
to explore novel molecular mechanisms of cancer development.
These studies have shown that the hsa-miR-196a2
rs11614913 polymorphism occurs in different types of cancer, but
the results are generally controversial and inadequate. In
addition, the sample size in each study was relatively small, with
statistical power too low to detect the association between the
hsa-miR-196a2 polymorphism and cancer risk. To solve the problem of
inadequate statistical power and controversial results, it is
necessary to carry out a systematic review and meta-analysis to
improve our current understanding of the association of the
hsa-miR-196a2 rs11614913 polymorphism with human cancer risk.
Subjects and methods
Search strategy
The databases, including PubMed and Embase, were
searched using: (miR-196a2[All Fields] OR rs11614913[All Fields])
AND (‘neoplasms’[MeSH Terms] OR ‘neoplasms’[All Fields] OR
‘cancer’[All Fields]). The search was restricted to case-control
studies published in English and Chinese updated to July 1, 2011.
All of the searched studies were reviewed and a manual search of
citations from the original studies was performed to identify
additional relevant articles.
Inclusion and exclusion criteria
Study quality was assessed according to the proposed
checklist of Little et al (35) for reporting and appraising studies
of genotype prevalence and gene-disease associations. A study in
which all or most of the criteria specified are satisfied would be
graded as high quality. The specified inclusion criteria for this
meta-analysis were the following: i) case-control studies: cases
were patients newly diagnosed and histopathologically confirmed
with different types of cancer, while controls were without cancer;
ii) evaluation of the association between the rs11614913 and cancer
risks; iii) correct statistical analysis and sufficient published
data for estimating odds ratio (OR) with 95% confidence interval
(CI); and iv) Hardy-Weinberg equilibrium (HWE). Animal studies,
pure cell studies, studies not concerned with cancer risk, repeated
or overlapping studies, studies without complete rs11614913
polymorphism distribution data and studies not fit for HWE were
excluded.
Data extraction
Two investigators independently extracted data using
standardized forms. When an agreement was not reached, a third
investigator resolved the conflict.
The following characteristics were extracted from
each study if available: i) first name of the author; ii)
publication year; iii) country or region of origin; iv) ethnicity
(different ethnic descents were categorized as Caucasian and
Asian); v) genotyping method; vi) source of control (population- or
hospital-based controls); vii) cancer type; viii) numbers of cases
and controls with miR-196a2 rs11614913 CC, CT and TT genotypes,
respectively; and ix) P-value for HWE.
Statistical analysis
Based on the complete hsa-miR-196a2 rs11614913
polymorphism distribution data in cases and controls, the crude ORs
with their 95% CIs were performed and displayed as forest plots to
assess the strength of association between the hsa-miR-196a2
rs11614913 polymorphism and susceptibility to cancer. The pooled
ORs were calculated for homozygote comparison (CC vs. TT),
heterozygote comparison (CC vs. CT), dominant model (CC vs. CT/TT)
and recessive model (CC/CT vs. TT), respectively. The significance
of the pooled OR was determined by the Z-test, and P<0.05 was
considered to denote statistical significance. Subgroup analyses
were performed for specific cancer types, genotypes, control
sources and ethnicities.
Heterogeneity of the study was explored by using
both Cochran Q statistic and estimating I2 test
(36). When the presence of
heterogeneity was detected (P-value <0.10 for the Q-test,
I2 values >50%), the random effects model
(DerSimonian Laird) was chosen. Otherwise, the fixed effects model
(Mantel-Haenszel method) was appropriately used to calculate the
pooled OR. HWE in the control group was assessed by the Chi-square
test for goodness of fit using a web-based program (http://ihg.gsf.de/cgi-bin/hw/hwa1.pl);
P<0.05 was considered significant.
Sensitivity analysis was carried out to assess the
stability of the results. A single study involved in the
meta-analysis was deleted each time to reflect the influence of the
ORs. Publication bias was assessed using Begg and Egger's formal
statistical test (statistical significance was defined as
P<0.10) (37,38). Statistical analyses were conducted
with Stata 10.0 (Stata Corp., College Station, TX, USA), using
two-sided P-values. Meta-analysis was performed using the ‘metan’
and ‘metabias’ STATA command.
Results
Characteristics of the included
studies
A total of 55 studies were considered to be relevant
by literature search from the PubMed and Embase databases (Fig. 1). Thirty studies were excluded by
article review, including 17 studies repeated or overlapped in the
PubMed and Embase databases; 6 studies were not concerned with
cancer risk research, 1 study was an animal study, 1 study was a
cell study, and 5 studies were meta-analysis. During the reading of
the 25 full-text manuscripts, 4 studies were excluded due to
incomplete rs11614913 polymorphism distribution data required for
OR calculation (10–13). For the remaining 21 records,
baseline characteristics of the patients and control subjects were
summarized, the study quality was assessed, HWE in particular was
assessed by Chi-square test; two records involving Indian
populations published in 2011 were excluded for disagreement with
HWE (P<0.05) (14,15), thus leaving 19 articles identified
with criteria for inclusion and exclusion (16–34).
All studies were case-control studies. The cases were patients with
cancer, and the controls were without cancer. The reported age and
gender distributions were also recorded. By quality assessment,
these studies satisfied most of the criteria specified by Little
et al (35). In the study
of Catucci et al (20), the
genotype frequencies were presented separately for a German and an
Italian group, thus each group in the study was considered
separately for meta-analysis. Therefore, a total of 20 studies
including 11,004 cases and 13,693 controls were included in the
meta-analysis.
Table I shows the
characteristics of the 20 studies, including first name of the
author, year of publication, country of origin, ethnicity,
genotyping method, source of controls, cancer type, numbers of
cases and controls with miR-196a2 rs11614913 CC, CT and TT
genotypes, respectively and P-value for HWE. Among 20 studies, 2
studies were published in 2009, 12 studies were published in 2010,
and 6 studies were published in 2011. There were 15 studies of
Asians, 4 studies of Caucasians, and 1 study of mixed population
with no detailed data on ethnicity. Multiple genotyping methods
were employed in the studies included in our analysis: 10 studies
using polymerase chain reaction-restriction fragment length
polymorphism (PCR-RFLP), 3 studies using TaqMan SNP genotyping
assay, 3 studies using polymerase chain reaction-ligation detection
reaction (PCR-LDR), others using MassARRAY multiplex, and DNA
sequencing. A blood sample was used for genotyping in all the
studies. There were 4 breast cancer studies, 4 hepatocellular
carcinoma (HCC) studies, 3 lung cancer studies, and other cancer
types. The controls of 16 studies mainly came from a hospital-based
healthy population (HB) matched for gender and age, and 4 studies
had population-based controls (PB). The distribution of genotypes
in the controls of all of the studies was in agreement with HWE
(P>0.05).
 | Table I.Characteristics of the 20 studies
included in the meta-analysis. |
Table I.
Characteristics of the 20 studies
included in the meta-analysis.
| | | | | | | Cases
| Controls
| P-valueHWE |
|---|
| Study (ref.) | Year | Country | Ethnicity | Genotyping
method | Cancer type | Control | CC | CT | TT | CC | CT | TT |
|---|
| Hu et al
(16) | 2009 | China | Asian | PCR-RFLP | Breast | PB | 239 | 483 | 287 | 218 | 517 | 359 | 0.21 |
| Hoffman et
al (17) | 2009 | USA | Mixed | MassARRAY | Breast | HB | 181 | 209 | 36 | 166 | 229 | 71 | 0.58 |
| Tian et al
(18) | 2010 | China | Asian | PCR-RFLP | Lung | PB | 253 | 512 | 293 | 209 | 519 | 307 | 0.70 |
| Peng et al
(19) | 2010 | China | Asian | PCR-RFLP | Gastric | HB | 76 | 94 | 43 | 56 | 107 | 50 | 0.94 |
| Catucci et
al (20) | 2010 | Germany | Caucasian | TaqMan | Breast | HB | 432 | 512 | 157 | 584 | 696 | 216 | 0.71 |
| Catucci et
al (20) | 2010 | Italy | Caucasian | TaqMan | Breast | HB | 334 | 330 | 87 | 532 | 550 | 161 | 0.32 |
| Dou et al
(21) | 2010 | China | Asian | PCR-LDR | Glioma | HB | 111 | 343 | 189 | 143 | 305 | 208 | 0.12 |
| Kim et al
(22) | 2010 | Korea | Asian | Fluorescence | Lung | HB | 187 | 305 | 162 | 155 | 300 | 185 | 0.13 |
| Li et al
(23) | 2010 | China | Asian | PCR-RFLP | HCC | HB | 78 | 150 | 82 | 42 | 102 | 78 | 0.40 |
| Liu et al
(24) | 2010 | USA | Caucasian | PCR-RFLP | HNSCC | HB | 350 | 565 | 194 | 383 | 545 | 202 | 0.74 |
| Okubo et al
(25) | 2010 | Japan | Asian | PCR-RFLP | Gastric | HB | 105 | 281 | 166 | 124 | 350 | 223 | 0.51 |
| Qi et al
(26) | 2010 | China | Asian | PCR-LDR | HCC | HB | 82 | 179 | 100 | 125 | 302 | 161 | 0.45 |
| Srivastava et
al (27) | 2010 | India | Asian | PCR-RFLP | Gallbladder | PB | 119 | 95 | 16 | 136 | 75 | 19 | 0.07 |
| Wang et al
(28) | 2010 | China | Asian | Snapshot | EC | HB | 148 | 262 | 48 | 128 | 250 | 111 | 0.60 |
| Chen et al
(29) | 2011 | China | Asian | PCR-LDR | CRC | HB | 27 | 64 | 35 | 94 | 206 | 107 | 0.79 |
| Hong et al
(30) | 2011 | Korea | Asian | TaqMan | Lung | HB | 86 | 224 | 96 | 96 | 198 | 134 | 0.16 |
| Zhan et al
(31) | 2011 | China | Asian | PCR-RFLP | CRC | HB | 68 | 128 | 56 | 113 | 267 | 163 | 0.85 |
| Zhou et al
(32) | 2011 | China | Asian | PCR-RFLP | CDCC | HB | 46 | 123 | 57 | 58 | 169 | 82 | 0.08 |
| Zhang et al
(33) | 2011 | China | Asian | PIRA-PCR | HCC | PB | 208 | 449 | 277 | 328 | 817 | 477 | 0.52 |
| Akkız et al
(34) | 2011 | Turkey | Caucasian | PCR-RFLP | HCC | HB | 77 | 86 | 22 | 58 | 87 | 40 | 0.49 |
Main meta-analysis results
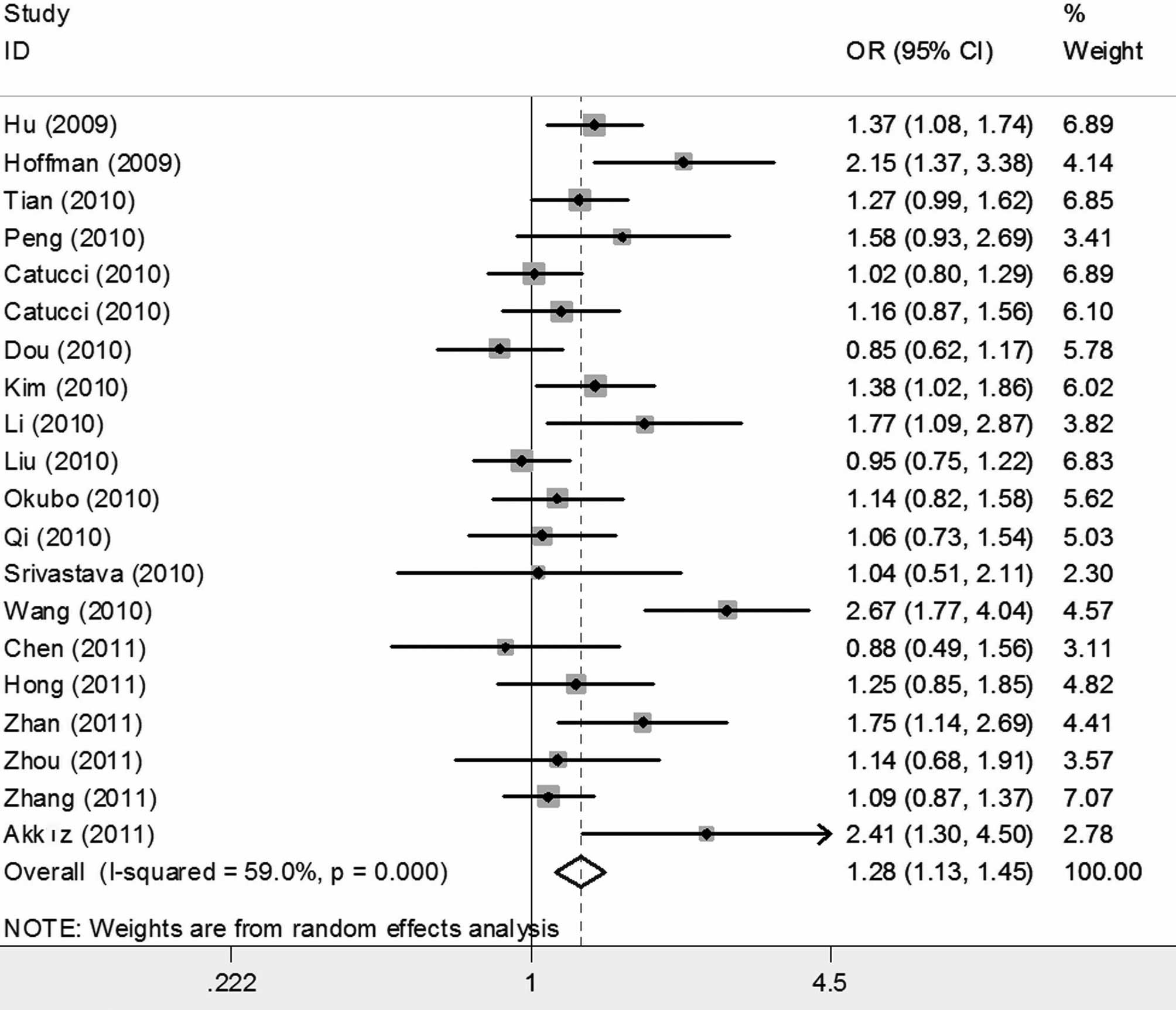
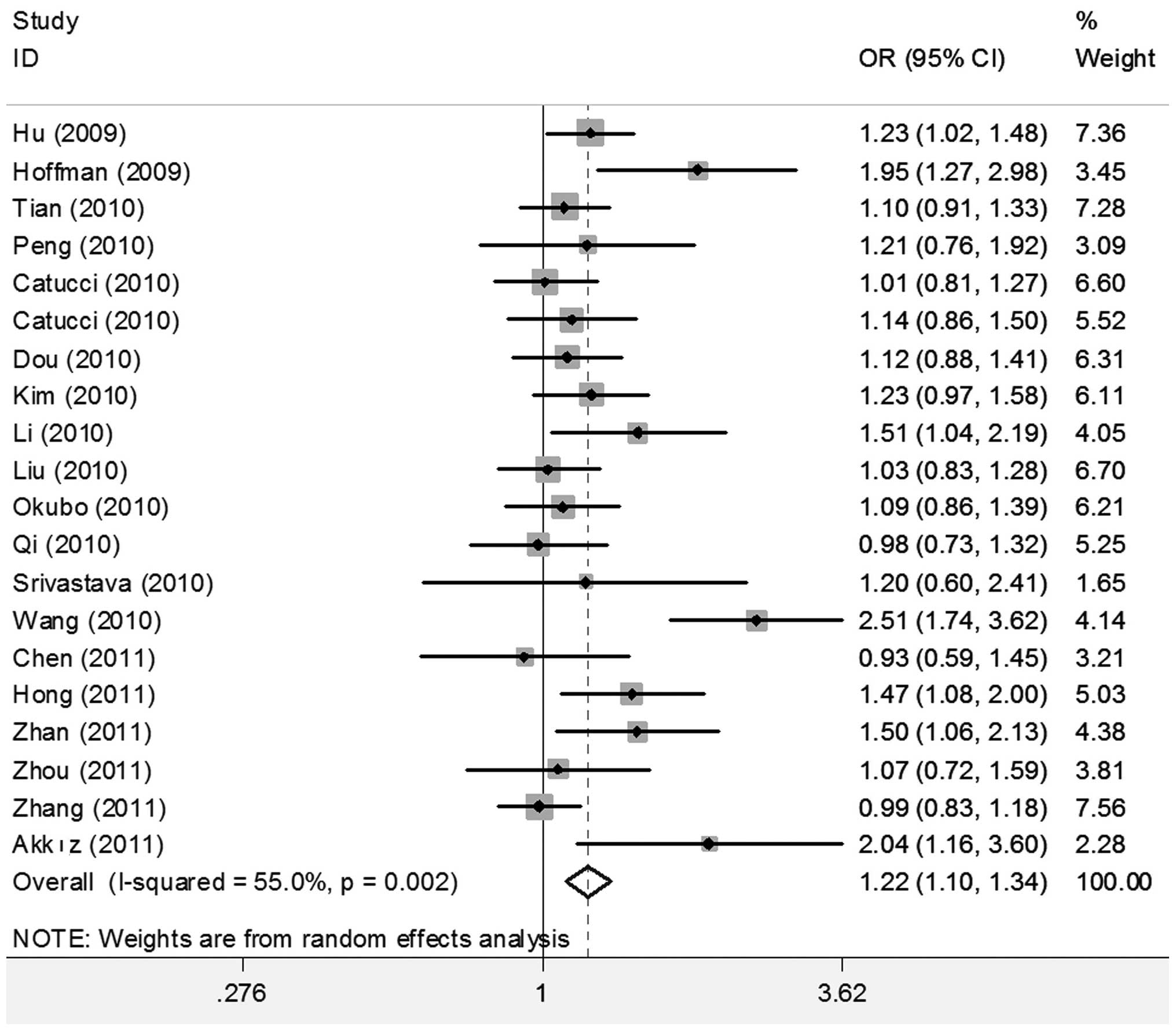
When all studies were pooled into the meta-analysis,
the hsa-miR-196a2 rs11614913 polymorphism was significantly
associated with an increased cancer risk in all genetic models (CC
vs. TT: OR=1.280; 95% CI 1.131–1.449, P<0.001; CT vs. TT:
OR=1.187, 95% CI 1.079–1.306, P<0.001; CC/CT vs. TT: OR=1.216;
95% CI 1.104–1.341, P<0.001; and CC vs. CT/TT: OR=1.115, 95% CI
1.025–1.213, P=0.011) (Figs. 2 and
3).
Next we performed the subgroup analysis of different
specific cancer types, genotypes, control sources and ethnicities
(Table II). In the different
cancer types, individuals carrying the CC genotype had an elevated
risk of breast cancer (CC vs. TT: OR=1.305, 95% CI 1.012–1.684,
P=0.041; and CC vs. CT/TT: OR=1.114, 95% CI 1.011–1.227, P=0.029),
lung cancer (CC vs. TT: OR=1.299, 95% CI 1.096–1.540, P=0.003; and
CC vs. CT/TT: OR=1.791, 95% CI 1.022–1.360, P=0.024), digestive
system cancer including gastric and CRC (CC vs. TT: OR=1.292, 95%
CI 1.041–1.603, P=0.020; and CC vs. CT/TT: OR=1.215, 95% CI
1.015–1.455, P=0.034) and HCC (CC vs. CT/ TT: OR=1.200, 95% CI
1.038–1.387, P=0.014) compared with those with the TT or TC/TT
genotypes. In addition, individuals carrying the CT genotype had an
elevated risk for breast cancer (CT vs. TT: OR=1.151, 95% CI
1.012–1.310, P=0.032) and other cancers (CT vs. TT: OR=1.352, 95%
CI 1.008–1.814, P=0.044) compared with those with the TT genotype.
Individuals carrying the CC/ CT genotype had an elevated risk of
lung cancer (CC/CT vs. TT: OR=1.206, 95% CI 1.054–1.380, P=0.007)
compared with those with the TT genotype.
| Table II.Subgroup meta-analysis results of the
hsa-miR-196a2 rs11614913 polymorphism and cancer risk. |
Table II.
Subgroup meta-analysis results of the
hsa-miR-196a2 rs11614913 polymorphism and cancer risk.
| Variables | n | CC vs. TT OR (95%
CI) | P-H | P-z | CT vs. TT OR (95%
CI) | P-H | P-z | CC+CT vs. TT OR
(95% CI) | P-H | P-z | CC vs. CT+TT OR
(95% CI) | P-H | P-z |
|---|
| Cancer types | | | | | | | | | | | | | |
| Breast | 4 | 1.305
(1.012–1.684) | 0.028 | 0.041 | 1.151
(1.012–1.310) | 0.161 | 0.032 | 1.223
(0.998–1.499) | 0.061 | 0.052 | 1.114
(1.011–1.227) | 0.208 | 0.029 |
| HCC | 4 | 1.374
(0.985–1.915) | 0.040 | 0.061 | 1.120
(0.873–1.438) | 0.087 | 0.373 | 1.215
(0.918–1.609) | 0.026 | 0.174 | 1.200
(1.038–1.387) | 0.407 | 0.014 |
| Gastro | 4 | 1.292
(1.041–1.603) | 0.187 | 0.020 | 1.118
(0.936–1.337) | 0.564 | 0.219 | 1.170
(0.988–1.385) | 0.346 | 0.068 | 1.215
(1.015–1.455) | 0.259 | 0.034 |
| Lung | 3 | 1.299
(1.096–1.540) | 0.895 | 0.003 | 1.204
(0.956–1.516) | 0.094 | 0.114 | 1.206
(1.054–1.380) | 0.281 | 0.007 | 1.171
(1.022–1.360) | 0.289 | 0.024 |
| Others | 5 | 1.203
(0.798–1.815) | 0.000 | 0.377 | 1.352
(1.008–1.814) | 0.007 | 0.044 | 1.289
(0.939–1.769) | 0.001 | 0.116 | 0.939
(0.756–1.166) | 0.024 | 0.567 |
| Genotyping | | | | | | | | | | | | | |
| PCR-RFLP | 10 | 1.318
(1.127–1.541) | 0.083 | 0.001 | 1.140
(1.038–1.252) | 0.674 | 0.006 | 1.181
(1.081–1.290) | 0.352 | 0.000 | 1.170
(1.014–1.350) | 0.015 | 0.032 |
| PCR-LDR | 3 | 0.925
(0.740–1.155) | 0.685 | 0.491 | 1.091
(0.911–1.307) | 0.364 | 0.341 | 1.041
(0.878–1.235) | 0.696 | 0.641 | 0.884
(0.730–1.070) | 0.214 | 0.206 |
| TaqMan | 3 | 1.104
(0.933–1.306) | 0.624 | 0.249 | 1.189
(0.922–1.533) | 0.088 | 0.182 | 1.148
(0.987–1.336) | 0.156 | 0.073 | 1.022
(0.913–1.144) | 0.743 | 0.708 |
| Others | 4 | 1.662
(1.113–2.482) | 0.001 | 0.013 | 1.437
(0.955–2.161) | 0.000 | 0.082 | 1.518
(1.012–2.278) | 0.000 | 0.044 | 1.237
(1.097–1.395) | 0.687 | 0.001 |
| Controls | | | | | | | | | | | | | |
| PB | 4 | 1.225
(1.071–1.401) | 0.551 | 0.003 | 1.051
(0.940–1.175) | 0.355 | 0.385 | 1.100
(0.990–1.221) | 0.416 | 0.076 | 1.120
(0.943–1.331) | 0.086 | 0.197 |
| HB | 16 | 1.314
(1.117–1.544) | 0.000 | 0.001 | 1.233
(1.097–1.386) | 0.013 | 0.000 | 1.361
(1.114–1.428) | 0.001 | 0.000 | 1.115
(1.010–1.231) | 0.017 | 0.031 |
| Ethnicity | | | | | | | | | | | | | |
| Asian | 15 | 1.283
(1.120–1.470) | 0.011 | 0.000 | 1.187
(1.058–1.331) | 0.010 | 0.003 | 1.214
(1.088–1.354) | 0.008 | 0.001 | 1.129
(1.019–1.251) | 0.037 | 0.021 |
| Caucasian | 5 | 1.308
(0.972–1.761) | 0.003 | 0.076 | 1.151
(1.079–1.228) | 0.110 | 0.046 | 1.253
(0.988–1.590) | 0.016 | 0.063 | 1.086
(0.934–1.263) | 0.048 | 0.283 |
| Total | 20 | 1.280
(1.131–1.449) | 0.000 | 0.000 | 1.187
(1.079–1.306) | 0.009 | 0.000 | 1.216
(1.104–1.341) | 0.002 | 0.000 | 1.115
(1.025–1.213) | 0.010 | 0.011 |
In the different genotypes, the hsa-miR-196a2
rs11614913 polymorphism was associated with a significantly
increased cancer risk in all genetic models by PCR-RELP (CC vs. TT:
OR=1.318, 95% CI 1.127–1.541, P=0.001; CT vs. TT: OR=1.410, 95% CI
1.038–1.252, P=0.006; CC/CT vs. TT: OR=1.181, 95% CI 1.081–1.290,
P<0.001; and CC vs. CT/TT: OR=1.170, 95% CI 1.014–1.350,
P=0.032), and others (CC vs. TT: OR=1.662, 95% CI 1.113–2.482,
P=0.013; CC/CT vs. TT: OR=1.518, 95% CI 1.012–2.278, P=0.044; and
CC vs. CT/TT: OR=1.237, 95% CI 1.097–1.395, P=0.001), but no
significant associations were observed by PCR-LDR and TaqMan.
In Asian, but not Caucasian ethnicity, significantly
increased risks were observed in all genetic models (CC vs. TT:
OR=1.283, 95% CI 1.120–1.470, P<0.001; CT vs. TT: OR=1.187, 95%
CI 1,058–1.331, P=0.003; CC/CT vs. TT: OR=1.214, 95% CI
1.088–1.354, P=0.001; and CC vs. CT/TT: OR=1.129, 95% CI
1.019–1.251, P=0.021).
Hospital-based studies demonstrated a significantly
increased risk in all genetic models (CC vs. TT: OR=1.314, 95% CI
1.117–1.544, P=0.001; CT vs. TT: OR=1.233; 95% CI 1.097–1.386,
P<0.001; CC/CT vs. TT: OR=1.361, 95% CI 1.114–1.428, P<0.001;
and CC vs. CT/TT: OR=1.115, 95% CI 1.010–1.231, P=0.031).
Population-based studies demonstrated significantly increased risks
only for the CC genotype when compared with the TT genotype
(OR=1.225, 95% CI 1.071–1.401, P=0.003).
Sensitivity analysis and publication
bias
The overall results in the random model or fixed
model were similar. In addition, sensitivity analysis was also
carried out by deleting a single study in the meta-analysis each
time. The results showed that no individual study affected the
overall OR dominantly (data not shown). There was no evidence for
publication bias according to Begg’s (z=1.40, P=0.163) and Egger’s
tests (t=1.32, P=0.202) for CC vs. CT/TT.
Discussion
Principal findings
In the present meta-analysis including 11,004 cases
and 13,693 controls, we found that the hsa-miR-196a2 rs11614913
polymorphism was associated with significantly increased overall
cancer risk in all genetic models. This meta-analysis provides
compelling evidence that the hsa-miR-196a2 rs11614913 polymorphism
may play a crucial role in the development of cancer and may be
used as a candidate biomarker for cancer susceptibility. Moreover,
in our subgroup analysis, we indicated that individuals carrying
the CC genotype had a significantly elevated risk of breast cancer,
lung cancer, digestive system cancer (including gastric and CRC)
and HCC compared with those with TT or TC/TT genotypes, consistent
with the total results. Ryan et al (39) suggested that variations in miRNAs
may be related to the risk of cancer and reported that the
rs11614913 polymorphism located in the hsa-miR-196a2 3′ mature
sequence affects the maturation and may affect target mRNA. Cell
culture experiments also indicated that high hsa-miR-196a levels
could suppress the activities of various cancer-related genes, such
as ANXA1 (Annexin A1), suppression of which is well documented in
various cancer types (40,41). Li et al (23) conducted an analysis of rs11614913
genotypes and the expression of mature miR-196a. They found that
the expression level of hsa-miR-196a was significantly higher in CC
patients or patients carrying at least one C allele than in TT
patients. The CC homozygotes were associated with a statistically
significant increase in mature miR-196a. Therefore, the altered
expression patterns of miR-196a influence its potential targets and
may play a role in the regulatory processes that occur during
cancer development.
Over the past several years, a large number of
distinct genotyping approaches have been designed for SNP detection
and identification, typically involving the amplification of the
target DNA sequence and the detection of SNPs, but each genotyping
method has its merit and demerit. A proper technology platform
should be adopted according to the sample size and the amount of
SNPs (42). The results of
PCR-RFLP, PCR-LDR, TaqMan and other methods to detect the
hsa-miR-196a2 rs11614913 polymorphism may not be in full accord. We
found that the hsa-miR-196a2 rs11614913 polymorphism significantly
increased cancer risk in all genetic models using the PCR-RFLP
method, but not using the PCR-LDR method. Therefore, a great deal
of effort should be devoted to developing more accurate, rapid, and
cost-effective technologies for SNP analysis.
In the subgroup analysis of source of controls and
ethnicities, hospital-based and population-based studies
demonstrated significantly increased risks for the CC genotype
compared with the TT genotype. A population-based control can
better represent the population, but hospital-based controls are
more readily obtainable in research. In addition, significantly
increased risks were observed in an Asian but not in a Caucasian
ethnic population, suggesting potentially different mechanisms in
different populations according to different genetic background and
environment. More studies in other ethnic groups may be necessary
for further progress in this area.
Strengths and limitations of the
meta-analysis
Several limitations of this meta-analysis should be
mentioned. First, the meta-analysis was limited by a relatively
small number of available studies. It is difficult to perform
subgroup analysis for every type of cancer. Second, our analysis
was limited to Asian and Caucasian ethnicities, so it is uncertain
whether these results are generalizable to other populations.
Third, restriction to studies published in English or Chinese may
confer potential language bias. In addition, studies with no
statistically significant results often have less chance for
publication. It is still difficult to rule out potential
publication bias in the meta-analysis. To confirm the role of the
hsa-miR-196a2 rs11614913 polymorphism in cancer risk requires
further larger studies in different populations and in different
types of cancer.
In spite of these limitations, our meta-analysis had
several strengths. In genetic association studies, the sample size
and statistical power are often of particular importance. We
overcame the limitations of a single study involving a relative
small number of subjects and the limitation in the statistical
significance, as relatively more sufficient number of cases and
controls were pooled from different studies, which significantly
increased the statistical power of the analysis. In addition, this
meta-analysis not only assessed the total strength of association
between the hsa-miR-196a2 rs11614913 polymorphism and overall
cancer risk in all genetic models, but also further performed
subgroup analysis of different specific cancer types, genotypes and
control sources to assess the polymorphism with cancer risk.
In conclusion, this meta-analysis provides
compelling evidence that the hsa-miR-196a2 rs11614913 polymorphism
may play a crucial role in the development of cancer, and that
screening of patients for the hsa-miR-196a2 rs11614913 polymorphism
is clinically useful for the prediction and prevention of
cancer.
Abbreviations:
|
miRNAs
|
microRNAs
|
|
SNPs
|
single nucleotide polymorphisms
|
|
OR
|
odds ratio
|
|
CI
|
confidence interval
|
|
HWE
|
Hardy-Weinberg equilibrium
|
|
PCR-RFLP
|
polymerase chain reaction-restriction
fragment length polymorphism
|
|
PCR-LDR
|
polymerase chain reaction-ligation
detection reaction
|
|
HCC
|
hepatocellular carcinoma
|
|
HB
|
hospital-based healthy controls
|
|
PB
|
population-based controls
|
|
ANXA1
|
Annexin A1
|
Acknowledgements
This study was supported by grants
from the NCET-10-0919, National Natural Science Foundation (no.
30801324), the Foundation of Shandong Natural Science (nos.
ZR2009CL005 and ZR2009CQ033), and the Scientific Research
Foundation of Shandong Educational Committee (nos. J09LF11 and
J08LG15).
References
|
1.
|
RC LeeRL FeinbaumV AmbrosThe C.
elegans heterochronic gene lin encodes small RNAs with
antisense complementarity to linCell758438541993
|
|
2.
|
NC LauLP LimEG WeinsteinDP BartelAn
abundant class of tiny RNAs with probable regulatory roles in
Caenorhabditis
elegansScience294858862200110.1126/science.106506211679671
|
|
3.
|
LA MacfarlanePR MurphyMicroRNA:
biogenesis, function and role in cancerCurr
Genomics11537561201010.2174/13892021079317589521532838
|
|
4.
|
DP BartelMicroRNAs: genomics, biogenesis,
mechanism, and
functionCell116281297200410.1016/S0092-8674(04)00045-514744438
|
|
5.
|
W LiuSY MaoWY ZhuImpact of tiny miRNAs on
cancersWorld J
Gastroenterol13497502200710.3748/wjg.v13.i4.49717278213
|
|
6.
|
CD DavisSA RossEvidence for dietary
regulation of microRNA expression in cancer cellsNutr
Rev66477482200810.1111/j.1753-4887.2008.00080.x18667010
|
|
7.
|
LB GaoP BaiXM PanJ JiaLJ LiWB LiangM
TangLS ZhangYG WeiL ZhangThe association between two polymorphisms
in pre-miRNAs and breast cancer risk: a meta-analysisBreast Cancer
Res Treat125571574201110.1007/s10549-010-0993-x20640596
|
|
8.
|
M Lagos-QuintanaR RauhutJ MeyerA
BorkhardtT TuschlNew microRNAs from mouse and
humanRNA9175179200310.1261/rna.2146903
|
|
9.
|
Z HuJ ChenT TianX ZhouH GuL XuY ZengR
MiaoG JinH MaY ChenH ShenGenetic variants of miRNA sequences and
non-small cell lung cancer survivalJ Clin
Invest11826002608200818521189
|
|
10.
|
H YangCP DinneyY YeY ZhuHB GrossmanX
WuEvaluation of genetic variants in microRNA-related genes and risk
of bladder cancerCancer
Res6825302537200810.1158/0008-5472.CAN-07-599118381463
|
|
11.
|
BC ChristensenM Avissar-WhitingLG
OuelletRA ButlerHH NelsonMD McCleanCJ MarsitKT KelseyMature
microRNA sequence polymorphism in MIR196A2 is associated with risk
and prognosis of head and neck cancerClin Cancer
Res1637133720201010.1158/1078-0432.CCR-10-065720501619
|
|
12.
|
Y YeKK WangJ GuH YangJ LinJA AjaniX
WuGenetic variations in microRNA-related genes are novel
susceptibility loci for esophageal cancer riskCancer Prev Res
(Phila)1460469200810.1158/1940-6207.CAPR-08-013519138993
|
|
13.
|
Y HorikawaCG WoodH YangH ZhaoY YeJ GuJ
LinT HabuchiX WuSingle nucleotide polymorphisms of microRNA
machinery genes modify the risk of renal cell carcinomaClin Cancer
Res1479567962200810.1158/1078-0432.CCR-08-119919047128
|
|
14.
|
GP GeorgeR GangwarRK MandalSN SankhwarRD
MittalGenetic variation in microRNA genes and prostate cancer risk
in North Indian populationMol Biol
Rep3816091615201110.1007/s11033-010-0270-420842445
|
|
15.
|
RD MittalR GangwarGP GeorgeT MittalR
KapoorInvestigative role of pre-microRNAs in bladder cancer
patients: a case-control study in North IndiaDNA Cell
Biol30401406201110.1089/dna.2010.115921345130
|
|
16.
|
Z HuJ LiangZ WangT TianX ZhouJ ChenR MiaoY
WangX WangH ShenCommon genetic variants in pre-microRNAs were
associated with increased risk of breast cancer in Chinese womenHum
Mutat307984200910.1002/humu.2083718634034
|
|
17.
|
AE HoffmanT ZhengC YiD LeadererJ WeidhaasF
SlackY ZhangT ParanjapeY ZhuMicroRNA miR-196a-2 and breast cancer:
a genetic and epigenetic association study and functional
analysisCancer
Res6959705977200910.1158/0008-5472.CAN-09-023619567675
|
|
18.
|
T TianY ShuJ ChenZ HuL XuG JinJ LiangP
LiuX ZhouR MiaoH MaY ChenH ShenA functional genetic variant in
microRNA-196a2 is associated with increased susceptibility of lung
cancer in ChineseCancer Epidemiol Biomarkers
Prev1811831187200910.1158/1055-9965.EPI-08-081419293314
|
|
19.
|
S PengZ KuangC ShengY ZhangH XuQ
ChengAssociation of microRNA-196a-2 gene polymorphism with gastric
cancer risk in a Chinese populationDig Dis
Sci5522882293201010.1007/s10620-009-1007-x19834808
|
|
20.
|
I CatucciR YangP VerderioS PizzamiglioL
HeesenK HemminkiC SutterB WappenschmidtM DickN ArnoldP BugertD
NiederacherA MeindlRK SchmutzlerCC BartramF FicarazziL TizzoniD
ZaffaroniS ManoukianM BarileMA PierottiP RadiceB BurwinkelP
PeterlongoEvaluation of SNPs in miR-146a, miR196a2 and miR-499 as
low-penetrance alleles in German and Italian familial breast cancer
casesHum Mutat31E1052E1057201010.1002/humu.2114119847796
|
|
21.
|
T DouQ WuX ChenJ RibasX NiC TangF HuangL
ZhouD LuA polymorphism of microRNA196a genome region was associated
with decreased risk of glioma in Chinese populationJ Cancer Res
Clin Oncol13618531859201010.1007/s00432-010-0844-520229273
|
|
22.
|
MJ KimSS YooYY ChoiJY ParkA functional
polymorphism in the pre-microRNA-196a2 and the risk of lung cancer
in a Korean populationLung
Cancer69127129201010.1016/j.lungcan.2010.04.01520466450
|
|
23.
|
XD LiZG LiXX SongCF LiuA variant in
microRNA-196a2 is associated with susceptibility to hepatocellular
carcinoma in Chinese patients with
cirrhosisPathology42669673201010.3109/00313025.2010.52217521080878
|
|
24.
|
Z LiuG LiS WeiJ NiuAK El-NaggarEM SturgisQ
WeiGenetic variants in selected pre-microRNA genes and the risk of
squamous cell carcinoma of the head and
neckCancer11647534760201010.1002/cncr.2532320549817
|
|
25.
|
M OkuboT TaharaT ShibataH YamashitaM
NakamuraD YoshiokaJ YonemuraT IshizukaT ArisawaI HirataAssociation
between common genetic variants in pre-microRNAs and gastric cancer
risk in Japanese
PopulationHelicobacter15524531201010.1111/j.1523-5378.2010.00806.x21073609
|
|
26.
|
P QiTH DouL GengFG ZhouX GuH WangCF
GaoAssociation of a variant in MIR 196A2 with susceptibility to
hepatocellular carcinoma in male Chinese patients with chronic
hepatitis B virus infectionHum
Immunol71621626201010.1016/j.humimm.2010.02.01720188135
|
|
27.
|
K SrivastavaA SrivastavaB MittalCommon
genetic variants in premicroRNAs and risk of gallbladder cancer in
North Indian populationJ Hum
Genet55495499201010.1038/jhg.2010.5420520619
|
|
28.
|
K WangH GuoH HuG XiongX GuanJ LiX XuK
YangY BaiA functional variation in pre-microRNA-196a is associated
with susceptibility of esophageal squamous cell carcinoma risk in
Chinese
HanBiomarkers15614618201010.3109/1354750X.2010.50529920722507
|
|
29.
|
H ChenLY SunLL ChenHQ ZhengQF ZhangA
variant in microRNA-196a2 is not associated with susceptibility to
and progression of colorectal cancer in ChineseIntern Med
JJan172011(Epub ahead of print)10.1111/j.1445-59942011.02434.x
|
|
30.
|
YS HongHJ KangJY KwakBL ParkCH YouYM KimH
KimAssociation between microRNA196a2 rs11614913 genotypes and the
risk of non-small cell lung cancer in Korean populationJ Prev Med
Public Health44125230201110.3961/jpmph.2011.44.3.12521617338
|
|
31.
|
JF ZhanLH ChenZX ChenYW YuanGZ XieAM SunY
LiuA functional variant in microRNA-196a2 is associated with
susceptibility of colorectal cancer in a Chinese populationArch Med
Res42144148201110.1016/j.arcmed.2011.04.00121565628
|
|
32.
|
B ZhouK WangY WangM XiZ ZhangY SongL
ZhangCommon genetic polymorphisms in pre-microRNAs and risk of
cervical squamous cell carcinomaMol
Carcinog50499505201110.1002/mc.2074021319225
|
|
33.
|
XW ZhangSD PanYL FengJB LiuJ DongYX
ZhangJG ChenZB HuHB ShenRelationship between genetic polymorphism
in microRNAs precursor and genetic prediposition of hepatocellular
carcinomaZhonghua Yu Fang Yi Xue Za Zhi452392432011(In
Chinese).
|
|
34.
|
H AkkızS BayramA BekarE AkgöllüY UlgerA
functional polymorphism in pre-microRNA-196a-2 contributes to the
susceptibility of hepatocellular carcinoma in a Turkish population:
a case-control studyJ Viral Hepat18e399e407201121692953
|
|
35.
|
J LittleL BradleyMS BrayM ClyneJ DormanDL
EllsworthJ HansonM KhouryJ LauTR O’BrienN RothmanD StroupE TaioliD
ThomasH VainioS WacholderC WeinbergReporting, appraising, and
integrating data on genotype prevalence and gene-disease
associationsAm J
Epidemiol156300310200210.1093/oxfordjournals.aje.a00017912181099
|
|
36.
|
JP HigginsSG ThompsonJJ DeeksDG
AltmanMeasuring inconsistency in
meta-analysesBMJ327557560200310.1136/bmj.327.7414.55712958120
|
|
37.
|
CB BeggM MazumdarOperating characteristics
of a rank correlation test for publication
biasBiometrics5010881101199410.2307/25334467786990
|
|
38.
|
M EggerSG DaveyM SchneiderC MinderBias in
meta-analysis detected by a simple, graphical
testBMJ315629634199710.1136/bmj.315.7109.6299310563
|
|
39.
|
BM RyanAI RoblesCC HarrisGenetic variation
in microRNA networks: the implications for cancer researchNat Rev
Cancer10389402201010.1038/nrc286720495573
|
|
40.
|
R LuthraRR SinghMG LuthraYX LiC HannahAM
RomansBA BarkohSS ChenJ EnsorDM MaruRR BroaddusA RashidCT
AlbarracinMicroRNA-196a targets annexin A1: a microRNAmediated
mechanism of annexin A1 downregulation in
cancersOncogene2766676678200810.1038/onc.2008.25618663355
|
|
41.
|
D ShenF NooraieY ElshimaliV LonsberryJ HeS
BoseD ChiaD SeligsonHR ChangL GoodglickDecreased expression of
annexin A1 is correlated with breast cancer development and
progression as determined by a tissue microarray analysisHum
Pathol3715831591200610.1016/j.humpath.2006.06.00116949910
|
|
42.
|
S KimA MisraSNP genotyping: technologies
and biomedical applicationsAnnu Rev Biomed
Eng9289320200710.1146/annurev.bioeng.9.060906.152037
|