Introduction
Alzheimer’s disease (AD) is a neurodegenerative
disease that is characterized by dementia as the main clinical
feature. AD mainly affects the elderly and has become one of the
major fatal diseases. Typical pathological features of AD include
β-amyloid protein (Aβ) deposition in the brain that forms senile
plaques (SPs), neurofibrillary tangles (NFTs) and neuronal
apoptosis.
A variety of hypotheses exist for AD risk factors,
and the trace element neurotoxicity theory has drawn an increasing
amount of attention. Zinc (Zn) ions are capable of causing Aβ
aggregation through the linking of histidines (the 13th amino acid)
of adjacent Aβ molecules, and there is evidence that Zn ions play a
key role in the pathogenesis and pathological process of AD
(1–6). Cadmium (Cd) is a harmful trace
element that causes morphological and biochemical abnormalities of
the central nervous system (CNS), memory loss and mental
retardation (7). Concentrations of
Cd and Cd/Zn are significantly higher in the blood and hair of AD
patients than in healthy people (8,9).
However, the correlation between Cd and Aβ has seldom been
studied.
Aβ is central to the development of AD and has
become the focus of current research. Aβ levels are determined by
biosynthesis and enzymatic degradation. For biosynthesis, Aβ is
generated from its precursor, amyloid precursor protein (APP). APP
has two metabolic pathways, namely the α-secretase pathway and the
β-secretase pathway. Under physiological conditions, the majority
of APP is cleaved by α-secretase into soluble APPα (sAPPα) and a
transmembrane fragment (C83); sAPPα is further cleaved by
γ-secretase into P3 and AICD. The cleavage site of α-secretase is
located at the Aβ segment of APP, which prevents the generation of
Aβ with a complete molecular sequence. A small part of APP is
cleaved by β-secretase at the N-terminal of Aβ, which generates a
secretive soluble APP derivative (sAPPβ) and a transmembrane
component [the C-terminal fragment containing 99 amino acids
(C99)]. C99 is further cleaved by γ-secretase into Aβ and AICD
(10,11). For enzymatic degradation, neutral
endopeptidase (NEP) is a major Aβ-degrading enzyme, and a large
number of studies have shown that the levels of NEP and Aβ
deposition are negatively correlated. In healthy people, the
synthesis and degradation of Aβ is balanced, and a steady low level
of Aβ is maintained.
In the present study, we examined the toxic effects
of Cd on Aβ levels in APP/PS1 transgenic mice. Special learning
capacity was detected using the Morris water maze test. The change
in free Zn ion concentration in the mouse brain was detected using
autometallography (AMG) to study the effect of Cd on Zn. The number
and size of SPs in brains was detected using immunohistochemistry.
The change of Aβ1-42 level was detected using ELISA.
Changes in the protein expression of APP, α-secretase (ADAM10),
sAPPα and NEP were detected using western blotting. We aimed to
explore the metal ion metabolism in the AD brain and its
correlation with the pathophysiology of AD.
Materials and methods
Experimental animals
A total of 24 male APP/PS1 transgenic mice (3 months
old, weighing 25–27 g) were purchased from the Experimental Animal
Center of China Medical University. Mice were randomly divided into
two groups, a control group and a Cd treatment group, with 12 mice
each. CdCl2 (Cd 2.5 mg/kg.d−1) was added to
the drinking water of the Cd treatment group, and drinking water
was normal for the control group. The protocols in this study were
approved by the Institutional Review Board and the Animal Care and
Use Committee of China Medical University (Shenyang, China).
Morris water maze
Place navigation test
For 2 days prior to the experiments, mice were
familiarized with the water maze environment in the morning and
afternoon. For each training session, the mice were placed into the
water 4 times, from the southeast, northeast, southwest and
northwest. Formal experiments began on day 3 and lasted 4 days. A
quadrant was selected randomly, and the mice were placed into the
water along the wall with their back against the platform. Escape
latency was the time that the mice took to reach the platform. If
mice could not find the platform within 1 min, they were led to it
by the experimenter, and the latency was recorded as 60 sec. The
swimming trajectory and movement distance were also recorded to
judge the learning capacity of mice.
Spatial probe test
On the 5th day of the experiment, the platform was
withdrawn. A quadrant was selected randomly, and the mice were
placed into the water with their face toward the wall. The swimming
trajectory and the number of crossings of the original platform
within 1 min were recorded to judge the memory capacity of
mice.
Immunohistochemical staining
After behavioral testing, APP/PS1 transgenic mice
were decapitated. Their brains were quickly removed, and one-half
was rapidly placed into 4% paraformaldehyde for fixation and the
preparation of conventional paraffin sectioning. The other half was
used to separate the cortex and hippocampus, which was stored at
−80°C. Brain tissue paraffin sections of mice in each group were
dewaxed in xylene, washed with PBS, boiled using a microwave for
antigen retrieval, naturally cooled, washed with PBS, and incubated
with 5% BSA at room temperature for 1 h, followed by incubation
with a mouse Aβ antibody (Sigma, 1:5000) at 4°C overnight. The
sections were washed with 0.01 M PBS thoroughly, incubated with
biotin-labeled goat anti-mouse IgG at room temperature for 2 h,
washed with 0.05 M Tris-HCl thoroughly, and incubated with SABC at
room temperature for 1 h. DAB coloration was monitored under a
microscope. The sections were washed with distilled water to stop
the reaction and counterstained with hematoxylin. The sections were
dehydrated using conventional techniques, made transparent and
mounted. Each group included 6 mice. Five sections of the same part
of the brain per mouse were selected, and images were captured
under an optical microscope. The optical density value of positive
SPs in the hippocampus and cortex was analyzed using IPP 6.0
software and compared statistically.
ELISA
The Aβ1-42 level was detected using an
ELISA kit (Biosource International) according to the manufacturer’s
instructions. Brain tissues were lysed with 5 M guanidine
hydrochloride diluted with standard dilution buffer (1:50) and
centrifuged (12,000 x g, 4°C, 25 min). The supernatant was
collected. A total of 50 μl of standard or sample was added to each
well followed by 50 μl of antibody. The solution was incubated on a
shaker at room temperature for 3 h. The supernatant was removed,
and the plate was washed 4 times. Enzyme-conjugated secondary
antibody (100 μl) was added and incubated at room temperature for
30 min. The supernatant was removed, and the plate was washed 4
times. Substrate (100 μl) was added and incubated at room
temperature for 30 min. The stop solution (100 μl) was added, and
OD450 values were detected on a microplate reader. The
Aβ1-42 level was calculated according to the standard
curve.
AMG
Mice were decapitated, and their brains were quickly
removed. Fresh tissue slices approximately 2 mm thick were cut from
the middle part of the hippocampus. According to the modified AMG
protocol described by Danscher et al (18), the slices were immediately immersed
in phosphate buffer (pH 7.4) containing 0.1% sodium sulfide and 3%
glutaraldehyde, incubated on a shaker at 4°C for 3 days, and washed
with 0.1 M PBS for 10 min. The slices were immersed in a 30%
sucrose solution at 4°C until they sank to the bottom of the glass.
Frozen sections (30-μm thick) were prepared. The slices were placed
in a staining cylinder that contained metal developing incubation
buffer (60 ml gum arabic solution, 10 ml citrate buffer, 15 ml
hydroquinone solution and 15 ml silver emulsion solution),
incubated in a 26°C water bath for 60 min, and immersed in a 5%
sodium thiosulfate solution for 10 min to stop the reaction. The
sections were washed with deionized water, dehydrated gradually
with ethanol, made transparent with xylene, and mounted with
neutral gum. Each group included 6 mice. Five sections of the same
part of the brain per mouse were selected, and the images were
acquired under an optical microscope. The optical density value of
positive Zn ion plaques in the cortex was analyzed using IPP 6.0
software and compared statistically.
Western blotting
The cerebral cortex and hippocampus tissues of
APP/PS1 transgenic mice were weighed and cut into pieces using
small scissors on ice. A 5X volume of protein lysis buffer was
added, and the tissues were sonicated and lysed at 4°C overnight.
The samples were centrifuged at 4°C 12,000 rpm for 30 min, and the
supernatant was collected. The protein level was determined using
the Coomassie Brilliant Blue assay. Protein (60 μg/10 μl) was
loaded, and the electrophoresis was stopped when the bromophenol
blue reached the bottom of the gel. The protein was transferred to
film at 4°C at 45 V overnight. The membranes were incubated with
primary antibodies against ADAM10 (1:1000), sAPPα (1:500), NEP
(1:500) and GAPDH (1:12000) at room temperature for 2 h, washed
with TTBS 3 times for 10 min, incubated with horseradish peroxidase
(HRP)-conjugated secondary antibody (1:5000) at room temperature
for 2 h, and washed with TTBS 3 times for 10 min. ECL luminescence
was performed, and the resulting images were captured and analyzed
using a Bio-Rad gel image analyzer.
Statistical analysis
A T-test analysis of the data was performed using
SPSS 15.0 software, and the results were presented as the means ±
standard deviation (SD). P<0.05 was considered to indicate a
statistically significant difference.
Results
Morris water maze test
APP/PS1 transgenic mice displayed significant
behavioral symptoms of AD. To examine whether Cd affected the
behavioral change, we used the Morris water maze test to detect the
memory ability of these two groups of mice (9 months old). During
the place navigation test that was conducted over 4 days, the
search latency of these two groups of mice decreased. Compared to
the control group, the movement trajectory of the Cd treatment
group was mainly along the wall and away from the platform
(Fig. 1), and the search latency
and distance were longer. The number of crossings of the platform
was significantly reduced (Fig. 2,
p<0.01).
Aβ immunohistochemistry
The number and size of SPs in the cerebral cortex
and hippocampus increased significantly in the Cd treatment group
(Fig. 3), and the results of the
optical density analysis showed that the difference was
statistically significant (p<0.01).
Aβ1-42 levels in brains
We used an ELISA kit to further detect changes in
Aβ1-42 levels. The Aβ1-42 levels in the Cd
treatment group (94.32±2.83 pg/mg) increased significantly compared
to those in the control group (67.25±3.45 pg/mg) (p<0.01,
Fig. 4).
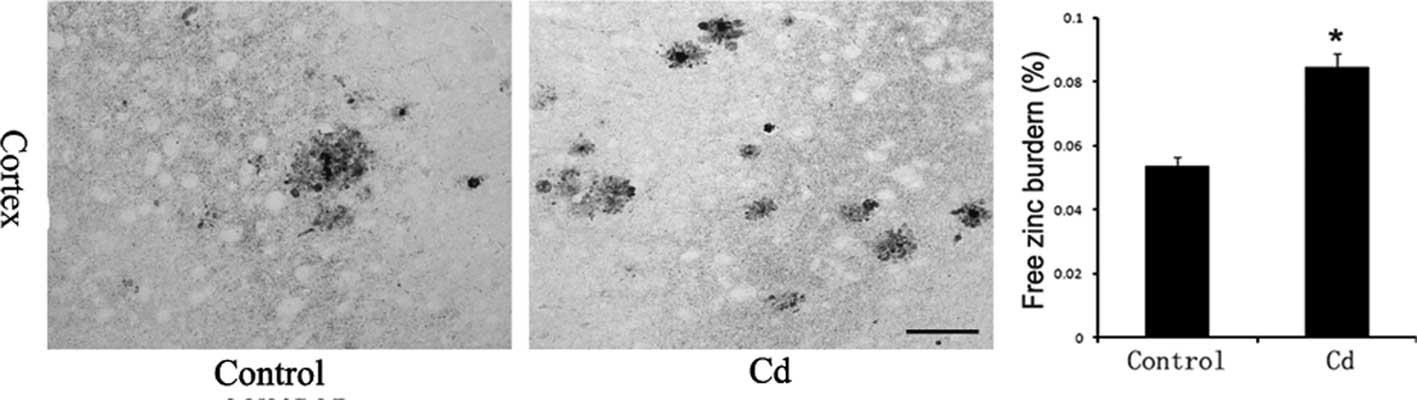
Free Zn ion levels in brains
Incubation with an AMG developer achieved the silver
amplification of Zn sulfide microcrystals that formed on slices,
which is highly specific and sensitive for the detection of free Zn
ions. The AMG-positive reaction product was brown, which indicates
the presence of Zn ions. Free Zn ion levels in the Cd treatment
group increased significantly compared to those in the control
group (p<0.01, Fig. 5).
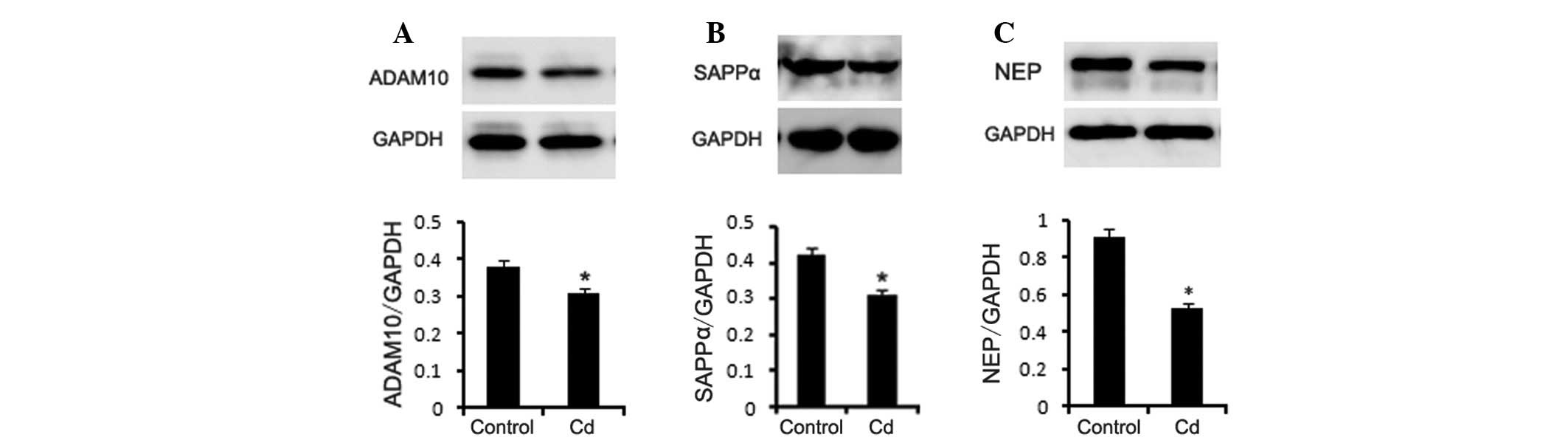
Western blotting
APP has two metabolic pathways, namely the
α-secretase pathway and the β-secretase pathway. The cleavage site
of α-secretase is located between the 16–17th amino acids of the Aβ
fragment of APP, which produces sAPPα and a transmembrane fragment
(C83) that prevents Aβ generation. In this study, we detected the
protein expression of ADAM10 (α-secretase) in the brains of the two
groups of mice using western blotting. The results showed that
ADAM10 and sAPPα protein levels were significantly lower in the Cd
treatment group (Fig. 6A–B,
p<0.01). NEP is an enzyme that is related to Aβ degradation. NEP
knockout or specific NEP inhibitors increase Aβ levels. Our results
indicated that the NEP protein level was decreased in the Cd
treatment group (Fig. 6C,
p<0.01).
Discussion
Cd has a number of biological toxicities, including
carcinogenesis, damage to kidney and bone, fetal toxicity and
teratogenic effects. In addition, Cd affects the nervous system
(12). Cd directly inhibits
thiol-containing enzymes; decreases the levels of norepinephrine,
serotonin, and acetylcholine; and has adverse effects on brain
metabolism (13). Exposure to Cd
decreases children’s IQ, visual development and learning ability
(7). Cd is also involved in the
formation of NFTs (14). Cd and
Cd/Zn are significantly higher in the blood and hair of AD patients
than in healthy people (8,9). Our results from the water maze test
revealed that the movement trajectory of the Cd treatment group was
mainly along the wall and away from the platform and that the
search latency and distance was longer, which indicated that Cd
treatment worsened the learning ability of the APP/PS1 transgenic
mice. The number of crossings of the platform was significantly
reduced in the Cd treatment group, which indicated that these mice
also had worse memory ability. Our results revealed that Cd
treatment aggravated the behavioral symptoms of AD.
Danscher et al confirmed that Zn ion levels
in the cerebral cortex and hippocampus of AD patients are
significantly higher than those in healthy people using atomic
absorption spectroscopy and X-ray microanalysis (15). This result suggests that Zn ions
play a key role in the pathogenesis and pathological processes of
AD. Over the years, in vitro experiments have shown that Zn
ions cause the fibrillar deposition of Aβ (16) and cause Aβ aggregation through the
linking of histidines (the 13th amino acid) of adjacent Aβ
molecules (17). In this study, we
applied the latest modified AMG technology (18) to quantitatively analyze the free Zn
ion distribution in the brains of two groups of mice. There were
AMG-positive SPs in the brains of APP/PS1 transgenic mice, which
indicated that there were numerous Zn ions in the SPs of Aβ
deposition. Moreover, free Zn ion levels were significantly higher
in the Cd treatment group. Immunohistochemistry results also
revealed that the number and size of SPs in the cerebral cortex and
hippocampus increased significantly in the Cd treatment group. The
above results indicated that Cd increased free Zn ion levels and
accelerated SP deposition in the brains of APP/ PS1 mice. Since Cd
is an anti-metabolite of Zn, we speculated that Cd may replace the
Zn ions in Zn enzymes, which would result in an increase in
extracellular Zn ions and an increase in SP deposition.
Brain Aβ was generated from its precursor, APP. APP
has two metabolic pathways, namely the α-secretase pathway and the
β-secretase pathway. Under physiological conditions, the majority
of APP is cleaved by α-secretase into sAPPα and a transmembrane
fragment (C83), and sAPPα is further cleaved by γ-secretase into P3
and AICD. The cleavage site of α-secretase is located in the Aβ
segment of APP, which prevents the generation of Aβ with a complete
molecular sequence. A very small part of APP is cleaved by
β-secretase at the N-terminal of Aβ, which generates a secretive
soluble APP derivative (sAPPβ) and a transmembrane component (C99).
C99 is further cleaved by γ-secretase into Aβ and AICD (10,11).
Aβ1-42 is highly cytotoxic (19). Our ELISA results confirmed that the
Aβ1-42 level was significantly higher in the Cd
treatment group. To study the possible mechanism of how Cd
increased the Aβ1-42 level, we used western blot
analysis to detect the protein expression of α-secretase (ADAM10)
and sAPPα. The results showed that the levels of these two proteins
were significantly lower in the Cd treatment group. ADAM10 has a
protective role in AD (20).
Therefore, we speculated that Cd inhibits the activity of
α-secretase, which leads to a greater metabolism of APP through the
β-secretase pathway and an increase in Aβ. Lower α-secretase
activity is associated with a reduced production of sAPPα, which
has neurotrophic and neuroprotective effects on neurons (21). A decrease in sAPPα could increase
the vulnerability of the surrounding neurons.
Several endopeptidases for Aβ degradation have been
found, including NEP, endothelin-converting enzyme and
insulin-degrading enzyme (22).
NEP is a major Aβ-degrading enzyme (23), and a large number of studies have
shown that the level of NEP and Aβ deposition is negatively
correlated. In healthy people, the synthesis and degradation of Aβ
is balanced, and a steady low level of Aβ is maintained.
NEP is a type II transmembrane glycoprotein in the
M13 Zn metalloproteinase family and is expressed at the presynaptic
membrane, axon and other neuronal parts. It is involved in the
degradation of enkephalins, bradykinin, substance P, somatostatin
and other neuropeptides (24). NEP
has 5 cleavage sites on Aβ and is capable of degrading monomers,
dimers and oligomers of Aβ1-40 and Aβ1-42
(24). NEP mainly degrades
extracellular Aβ1-42 (25). Many researchers have suggested that
NEP is an important Aβ-degrading enzyme in the brain; NEP
downregulation increases Aβ aggregation and its upregulation
reduces Aβ levels in the brain (25,26).
Our results revealed that NEP was significantly reduced in the Cd
treatment group, which indicates that Cd may reduce Aβ degradation
and increase Aβ aggregation through a reduction in NEP
expression.
In addition, Zn plays a role in the composition of
the active sites of more than 200 enzymes, including α-secretase
and NEP in vivo. Many of the toxic effects of Cd are related
to the complex interaction between Zn and Cd. Since Cd has a
stronger binding capacity with thiol, carboxyl and hydroxyl than
Zn, it is capable of replacing Zn in Zn enzymes. The replacement of
Zn with Cd inactivates these enzymes, which causes the dysfunction
of the human brain and triggers AD. The detailed mechanisms of AD
require further research.
Acknowledgements
We appreciate the valuable comments
from other members of our laboratories.
References
|
1
|
Jia Y, Jeng JM, Sensi SL and Weiss JH:
Zn2+ currents are mediated by calcium-permeable
AMPA/kainate channels in cultured murine hippocampal neurones. J
Physiol. 543:35–48. 2002.PubMed/NCBI
|
|
2
|
Koh JY and Choi DW: Zinc toxicity on
cultured cortical neurons: involvement of N-methyl-D-aspartate
receptors. Neuroscience. 60:1049–1057. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Palmiter RD and Findley SD: Cloning and
functional characterization of a mammalian zinc transporter that
confers resistance to zinc. EMBO J. 14:639–649. 1995.PubMed/NCBI
|
|
4
|
Rogers EE, Eide DJ and Guerinot ML:
Altered selectivity in an Arabidopsis metal transporter. Proc Natl
Acad Sci USA. 97:12356–12360. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vallee BL and Auld DS: Zinc coordination,
function, and structure of zinc enzymes and other proteins.
Biochemistry. 29:5647–5659. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vallee BL and Falchuk KH: The biochemical
basis of zinc physiology. Physiol Rev. 73:79–118. 1993.PubMed/NCBI
|
|
7
|
Wu X, Jin T, Wang Z, Ye T, Kong Q and
Nordberg G: Urinary calcium as a biomarker of renal dysfunction in
a general population exposed to cadmium. J Occup Environ Med.
43:898–904. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lui E, Fisman M, Wong C and Diaz F: Metals
and the liver in Alzheimer’s disease. An investigation of hepatic
zinc, copper, cadmium, and metallothionein. J Am Geriatr Soc.
38:633–639. 1990.
|
|
9
|
Panayi AE, Spyrou NM, Iversen BS, White MA
and Part P: Determination of cadmium and zinc in Alzheimer’s brain
tissue using inductively coupled plasma mass spectrometry. J Neurol
Sci. 195:1–10. 2002.
|
|
10
|
Lambert MP, Barlow AK, Chromy BA, et al:
Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent
central nervous system neurotoxins. Proc Natl Acad Sci USA.
95:6448–6453. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Torreilles F and Touchon J: Pathogenic
theories and intrathecal analysis of the sporadic form of
Alzheimer’s disease. Prog Neurobiol. 66:191–203. 2002.PubMed/NCBI
|
|
12
|
Bressler JP, Olivi L, Cheong JH, Kim Y,
Maerten A and Bannon D: Metal transporters in intestine and brain:
their involvement in metal-associated neurotoxicities. Hum Exp
Toxicol. 26:221–229. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jomova K and Valko M: Advances in
metal-induced oxidative stress and human disease. Toxicology.
283:65–87. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiang LF, Yao TM, Zhu ZL, Wang C and Ji
LN: Impacts of Cd(II) on the conformation and self-aggregation of
Alzheimer’s tau fragment corresponding to the third repeat of
microtubule-binding domain. Biochim Biophys Acta. 1774:1414–1421.
2007.PubMed/NCBI
|
|
15
|
Danscher G, Jensen KB, Frederickson CJ, et
al: Increased amount of zinc in the hippocampus and amygdala of
Alzheimer’s diseased brains: a proton-induced X-ray emission
spectroscopic analysis of cryostat sections from autopsy material.
J Neurosci Methods. 76:53–59. 1997.PubMed/NCBI
|
|
16
|
Bush AI, Pettingell WH, Multhaup G, et al:
Rapid induction of Alzheimer A beta amyloid formation by zinc.
Science. 265:1464–1467. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu ST, Howlett G and Barrow CJ:
Histidine-13 is a crucial residue in the zinc ion-induced
aggregation of the A beta peptide of Alzheimer’s disease.
Biochemistry. 38:9373–9378. 1999.PubMed/NCBI
|
|
18
|
Danscher G, Stoltenberg M, Bruhn M,
Sondergaard C and Jensen D: Immersion autometallography:
histochemical in situ capturing of zinc ions in catalytic
zinc-sulfur nanocrystals. J Histochem Cytochem. 52:1619–1625. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jung KM, Astarita G, Yasar S, et al: An
amyloid beta(42)-dependent deficit in anandamide mobilization is
associated with cognitive dysfunction in Alzheimer’s disease.
Neurobiol Aging. May 3–2011.(Epub ahead of print).
|
|
20
|
Endres K and Fahrenholz F: The role of the
anti-amyloidogenic secretase ADAM10 in shedding the APP-like
proteins. Curr Alzheimer Res. May 23–2011.(Epub ahead of
print).
|
|
21
|
Hook VY and Reisine TD: Cysteine proteases
are the major beta-secretase in the regulated secretory pathway
that provides most of the beta-amyloid in Alzheimer’s disease: role
of BACE 1 in the constitutive secretory pathway. J Neurosci Res.
74:393–405. 2003.PubMed/NCBI
|
|
22
|
Iwata N and Saido TC: Amyloid-beta peptide
metabolism and Alzheimer’s disease. Nihon Yakurigaku Zasshi.
122:5–14. 2003.
|
|
23
|
Leissring MA, Farris W, Chang AY, et al:
Enhanced proteolysis of beta-amyloid in APP transgenic mice
prevents plaque formation, secondary pathology, and premature
death. Neuron. 40:1087–1093. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Iwata N, Mizukami H, Shirotani K, et al:
Presynaptic localization of neprilysin contributes to efficient
clearance of amyloid-beta peptide in mouse brain. J Neurosci.
24:991–998. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Marr RA, Rockenstein E, Mukherjee A, et
al: Neprilysin gene transfer reduces human amyloid pathology in
transgenic mice. J Neurosci. 23:1992–1996. 2003.PubMed/NCBI
|
|
26
|
Sakai A, Ujike H, Nakata K, et al:
Association of the neprilysin gene with susceptibility to
late-onset Alzheimer’s disease. Dement Geriatr Cogn Disord.
17:164–169. 2004.PubMed/NCBI
|