Introduction
The reactivity of blood vessels depends on their
structure and the presence of calcium ions (1). It is modulated by numerous factors,
which activate specific signaling pathways leading to contraction
or relaxation of smooth muscle. The action of various
vasodilatation substances may be altered under the influence of
similar or quite different modulators. Vascular reactivity may vary
with age, which probably is associated with endothelial
dysfunction, leading to a reduction in nitric oxide synthesis
(2–4). Angiotensin II (ANG II) triggers
vasoconstriction via a metabotropic AT1 receptor (5). ANG II also regulates smooth muscle
cell (SMC) growth, has an effect on apoptosis and migration and has
proinflammatory action. In addition, it causes the production of
other growth- and contraction-stimulating factors. It is,
therefore, important both for maintaining the proper structure and
function of blood vessels and may mediate pathophysiological
processes leading to the development of cardiovascular diseases
(6). Phenylephrine (PHE) is an
agonist of the α1-adrenergic metabotropic receptor through which it
induces vasoconstriction (7).
Both substances, ANG II and PHE, act through G
proteins, which leads to stimulation of phospholipase C and the
synthesis of secondary messengers: IP3 and DAG (8–10).
IP3 binds to the endoplasmic reticulum membrane (ER)
IP3R receptors and causes the release of Ca2+
from intracellular pools. The ryanodin receptors (RyR), stimulated,
among others, by caffeine (1,11,12)
are an alternative way for the release of calcium from the ER.
Contraction of vascular smooth muscle may also occur via calcium
ions escaping from the extracellular space through channels in the
cell membrane [receptor-operated Ca2+ channels (ROC)]
activated by ligand ANG II or PHE (13).
Studies on vas deferens (human and rat) and rat tail
artery have shown that receptor associated G-protein modulation may
be influenced by sodium nitroprusside and 8Br-cGMP (14–16).
Nitric oxide derived from endothelium is a major vasodilatation
factor (17). Acetylcholine can
stimulate the release of nitric oxide in a cGMP-mediated relaxing
effect (18,19). However, studies using isolated
human placental villous arteries found that NO donors and 8Br-cGMP
did not cause relaxation of arteries contracted with caffeine. The
mechanism of nitric oxide action on the cardiac calcium release
channel (ryanodine receptor) (CRC) in canines was explored
(20,21). Lim et al discussed various
ways in which nitric oxide can modulate cardiac ryanodine receptor
function and suggested the possibility of pharmacological
strategies in heart failure, related to the considered mechanisms
(22).
The aim of this study was to assess the role of
acetylcholine and calcium ions in modulating the contraction
induced by ANG II, PHE and caffeine. These substances acted
together with the participation of calcium ions mobilized from
intracellular stores, but also (as in the case of ANG II and PHE)
using an extracellular pool of these ions.
Materials and methods
The study was performed on perfunded tail arteries
of male Wistar weighing 250–350 g, euthanized with an
intraperitoneally injection of urethane at the dose of 120 mg/kg.
The cannula was introduced in the proximal section of rat tail
artery (2.5–3 cm in length) and combined with a perfusion system
and a set that allows constant measurement and recording of
perfusion pressure. After loading the distal end of the isolated
artery with a weight of 500 mg, the preparation was placed upright
in a thermostated vessel for isolated organs 20 ml in volume and
oxygenated with physiological fluid at a temperature of 37°C.
Perfusion fluid flow was increased gradually to 1 ml/min.
The experiments were carried out to determine the
importance of intracellular and extracellular pools of
Ca2+ in reactions induced by ANG II (30 nM/l), PHE (3
μM/l) and caffeine (100 μM/l) in control conditions
and after addition of L-NNA (NOSe inhibitor) or ODQ (a soluble form
of GC inhibitor) and in the presence of increasing concentrations
of acetylcholine using two types of Krebs fluid: i) FPSS –
Ca2+-free EGTA-Krebs with the following composition:
NaCl (71.8 mM/l), KCl (4.7 mM/l), NaHCO3 (28.4 mM/l),
MgSO4 (2.4 mM/l), KH2PO4 (1.2
mM/l), glucose (11.1 mM/l) with the addition of EGTA (30
μM/l); ii) PSS – fluid with Ca2+ EGTA-Krebs
(normal) with the following composition: NaCl (71.8 mM/l), KCl (4.7
mM/l), CaCl2 (1.7 mM/l), NaHCO3 (28.4 mM/l),
MgSO4 (2.4 mM/l), KH2PO4 (1.2
mM/l), glucose (11.1 mM/l) with addition of EGTA (30 μM/l),
after emptying the intracellular pool of calcium ions.
The increase in pressure of the perfusate in the
experimental system was an exponent of vessel spasm. The Ethical
Committee for the Affairs of Experiments on Animals in Bydgoszcz
approved the protocol of the experiments undertaken (No.
1/2008-4).
Statistical analysis was performed by determining
the mean and standard deviation. Statistical differences were
evaluated by Student’s t-test. A p-value <0.05 was considered to
indicate a statistically significant difference.
Results
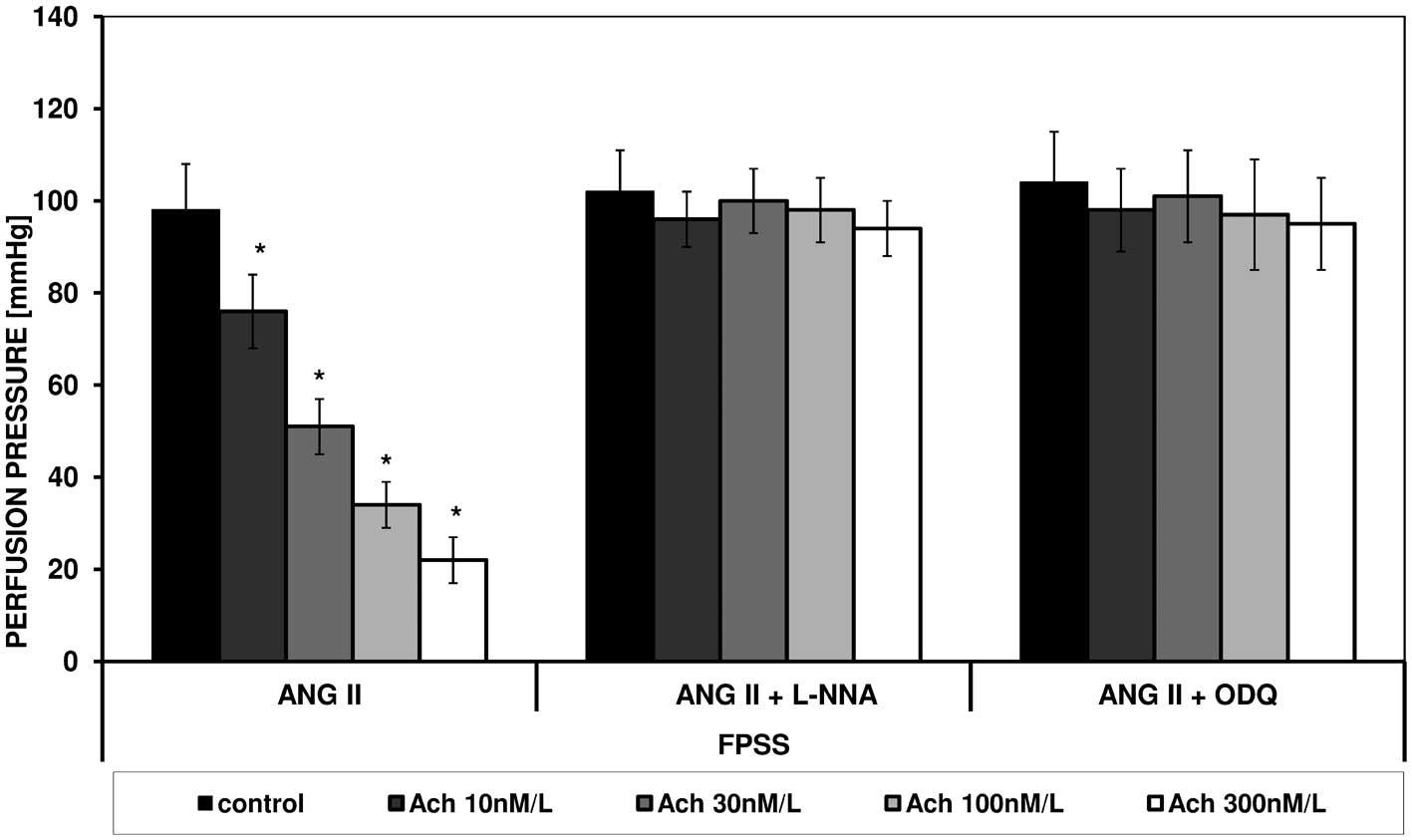
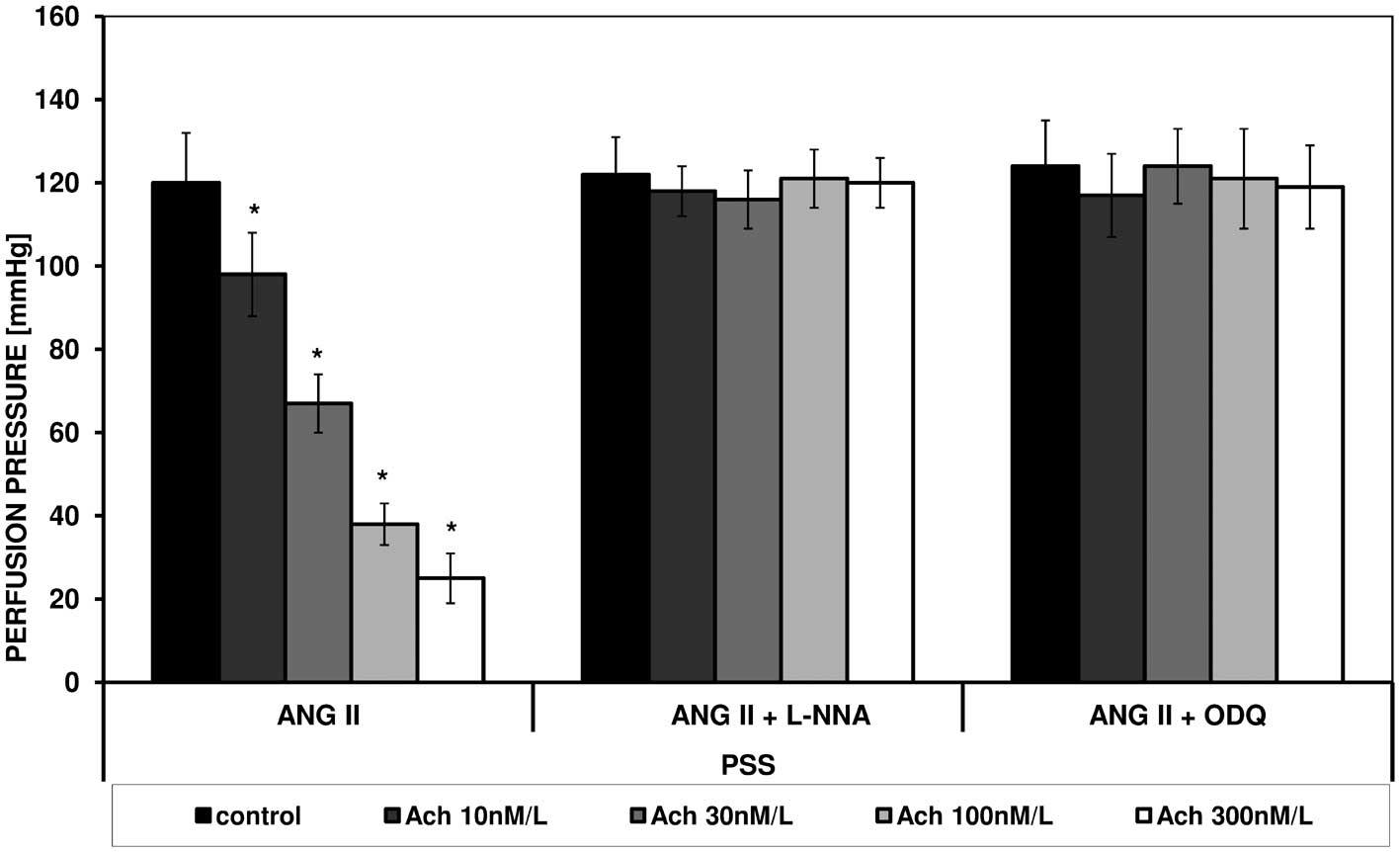
ANG II caused an increase in perfusion pressure in
FPSS and PSS. Under the influence of increasing concentrations of
acetylcholine a statistically significant reduction in perfusion
pressure in both types of fluid was noted (Figs. 1 and 2).
In the presence of L-NNA and ODQ in both solutions
no changes in contraction stimulated by ANG II or spasmolytic
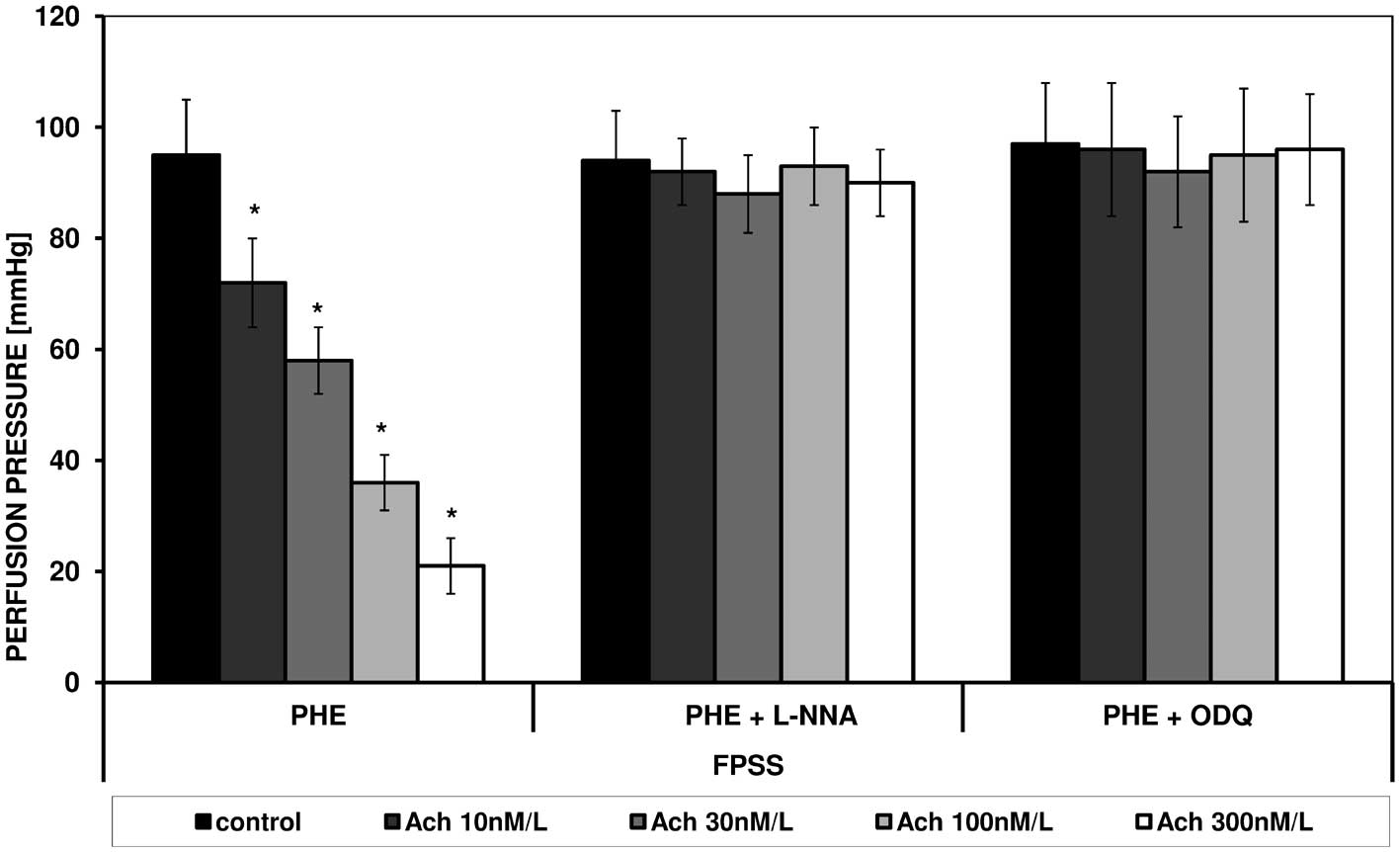
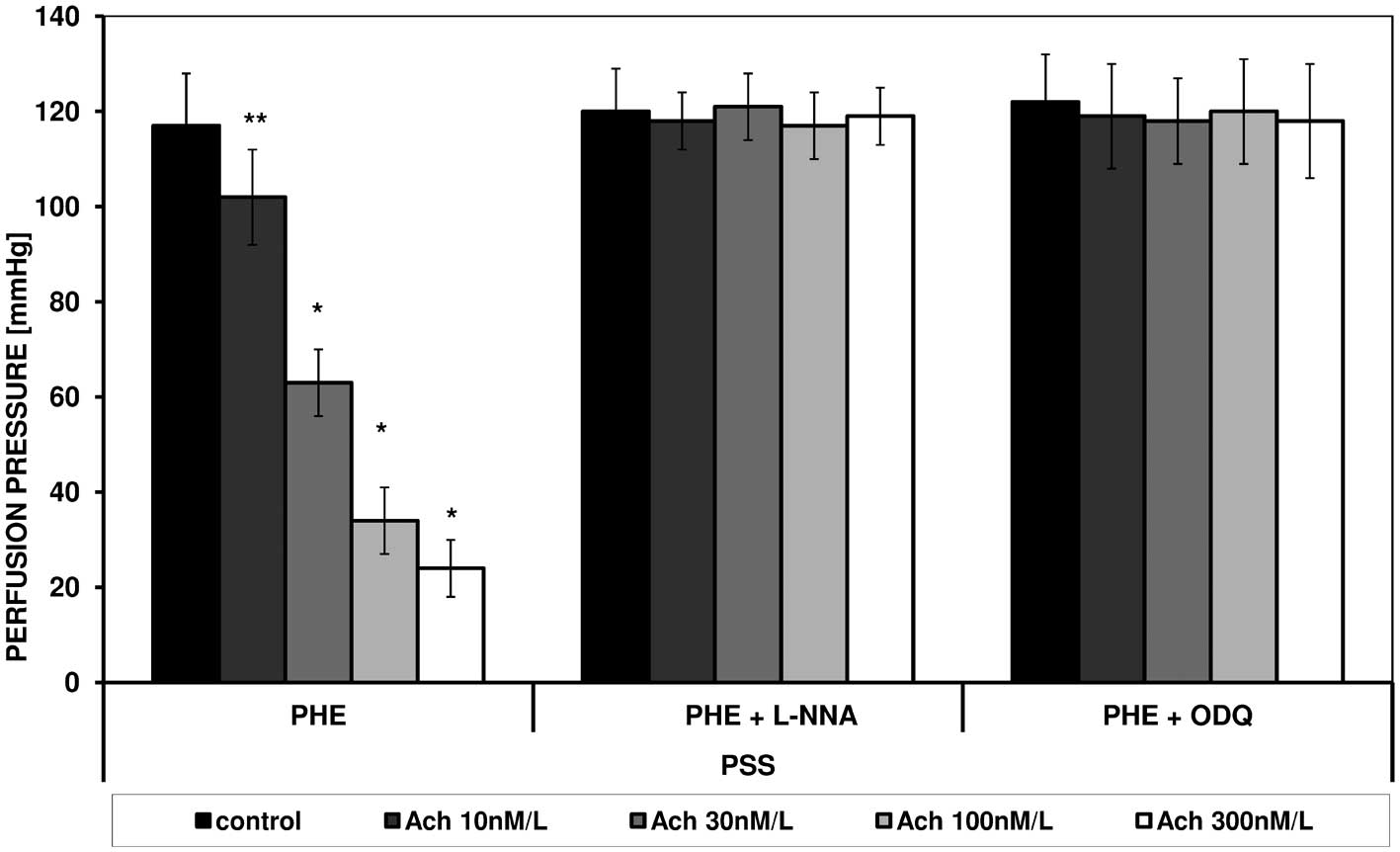
effect of acetylcholine were observed (Figs. 1 and 2). PHE, in a similar manner to ANG II
caused contraction in FPSS and PSS, which was similarly modulated
by acetylcholine. Figs. 3 and
4 present the effect of increasing
concentrations of acetylcholine on the perfusion pressure induced
by PHE in the presence of L-NNA and ODQ, respectively, in FPSS and
PSS.
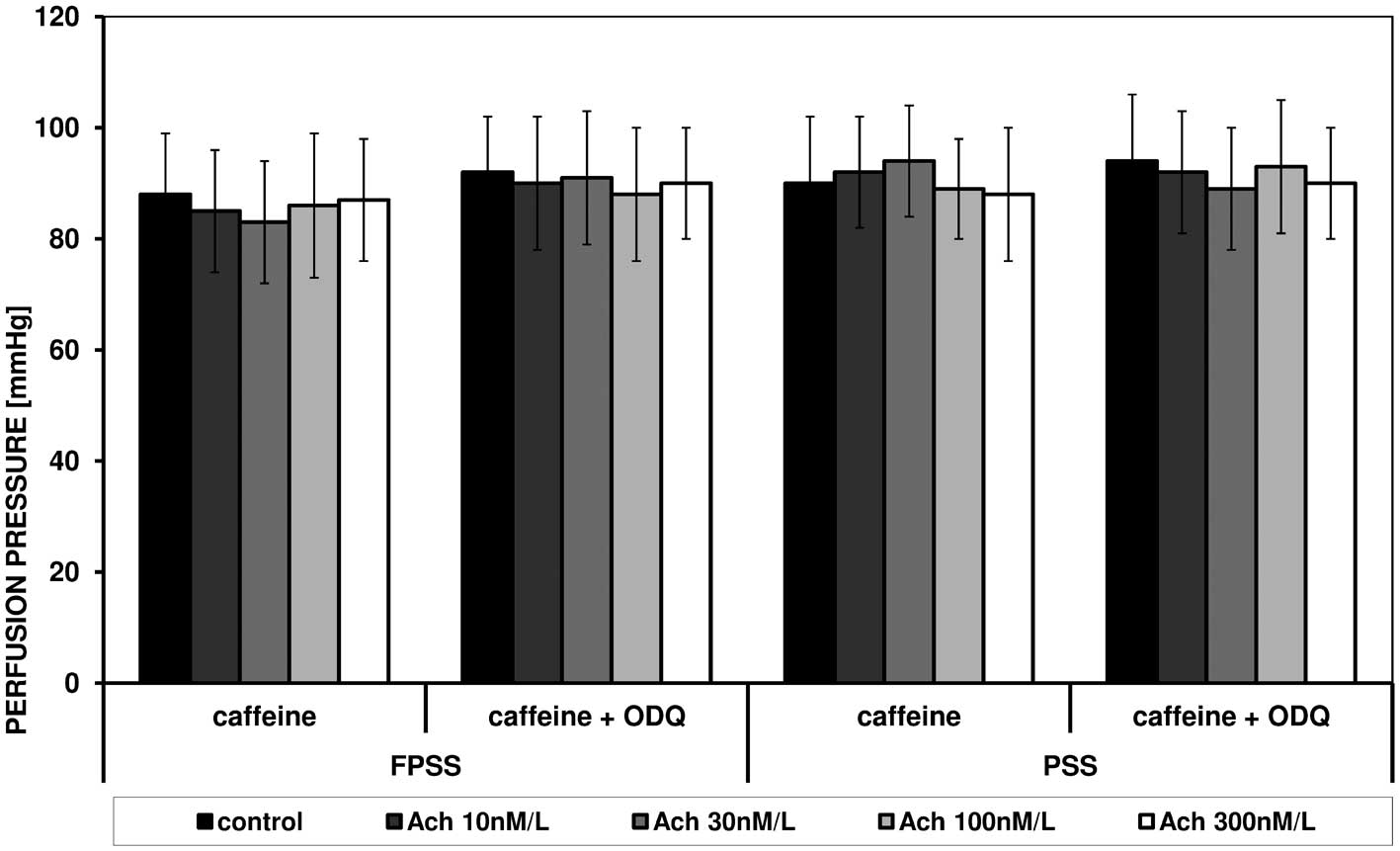
Caffeine induced an increase in perfusion pressure
in FPSS and PSS, and these reactions were not altered under the
influence of acetylcholine and ODQ. Fig. 5 shows the effect of increasing
concentrations of acetylcholine on caffeine-triggered contraction
in the presence of ODQ, FPSS and PSS.
Discussion
Calcium ions are an essential element in muscle
contraction. Vascular tone can be adjusted by a variety of
substances that stimulate the release of calcium from cellular
stores, that cause the influx of calcium from the outside, and that
stimulate sensitivity to calcium ions. A key role in regulating
muscle function is maintaining the concentration of calcium ions
within a very narrow range and regulating their ability for rapid
increase (1). An increase in
intracellular calcium levels precedes and induces the contraction
of smooth muscle. Acetylocholine decreases arterial tension due to
the release of NO from endothelial cells, thus it stimulates the
NO/cGMP signaling pathway (23).
Cyclic nucleotides, e.g., cAMP and cGMP, which regulate the
function of ion channels and calcium levels in the cell through the
appropriate protein kinases exhibit functional antagonism of
calcium ions in smooth muscle (24,25).
In experiments performed on mouse lung slices NO-induced relaxation
was enhanced by selective inhibitors of cGMP-specific
phosphodiesterase-5 (zaprinast or vardenafil), but was blocked by
ODQ and by Rp-8-pCPT-cGMPS, an inhibitor of protein kinase G. NOC-5
(nitric oxide donor), cGMP analogues and selective PKG activators
8Br-cGMP and 8pCPT-cGMP were found to induce airway relaxation and
decrease the frequency of Ca2+ oscillations (26). Studies using human mesenteric
arteries showed that guanylate cyclase activators modulate vascular
responses in conditions of ischemia/reperfusion (27). The aim of the present study was to
determine the role of acetylcholine and calcium ions in modulating
the vascular contraction induced by angiotensin II, phenylephrine
and caffeine.
The experiments were carried out in PSS (after
emptying the intracellular pool of calcium ions) and FPSS to
determine the importance of the extracellular and the intracellular
pools of calcium ions. Metabotropic receptor agonists, ANG II and
PHE, led to an increase in perfusion pressure in both types of
solutions; the reactions were stronger in PSS. Comparison of the
results indicates that the vascular contraction induced by the
studied substances was caused by the effect of calcium ions
released from the endoplasmic reticulum (via the IP3
receptor), entering the cell from the outside after opening the
appropriate channels in the membrane. Similar observations were
also derived from experiments on rat aorta and human mesenteric
arteries (28,29).
The existence of a contraction mechanism independent
of the intracellular calcium pool confirms previous experience
using rat tail artery and human mesenteric arteries with
xestospongin C, an IP3 receptor antagonist (29,30).
Acetylcholine reduced the vascular contraction stimulated by ANG II
and PHE in a concentration-dependent manner. Additionally, in
experiments using human mesenteric arteries such an effect of
acetylcholine was observed (18).
Ji et al in studies of rat aorta demonstrated that the
inhibitory effect of acetylcholine was associated with the presence
of endothelial cells and this effect was not present in experiments
carried out in arteries denuded of endothelium (27). Another series of studies found that
blocking NO synthase (after addition of L-NNA) and soluble guanyl
cyclase (GC, after addition of ODQ) led to the elimination of the
relaxing effects of acetylcholine. These results confirm the
dependence of acetylcholine on NO synthesis and activation of
GC.
A subsequent experiment was performed in FPSS and
PSS using caffeine, agonist of ryanodin receptors in the
endoplasmic reticulum, as a factor stimulating vascular
contraction. Previous studies in human mesenteric arteries showed
that emptying the intracellular pool of Ca2+ and
blockage of Ca2+-ATPase (by specifying thapsigargin)
caused the abolition of the response to caffeine (29). In contrast to the results of the
metabotropic receptor agonists, acetylcholine did not inhibit
caffeine-triggered contraction. This effect was consistent with
previous reports (28,29). It was previously shown that the
NPS, as a donor of NO, decreased rat artery contraction, induced by
ANG II and PHE, but was not affected by caffeine (27). The effect of NO on Ca2+
sensitivity of airway SMCs was examined in mouse lung slices
permeabilized to Ca2+ by treatment with caffeine and
ryanodine. Neither NOC-5 nor 8pCPT-cGMP induced relaxation in
agonist-contracted Ca2+-permeabilized airways (26). Slupski et al showed that
nitric oxide may reduce lung damage caused by increased vascular
resistance and arterial pressure after ischemia/reperfusion
(30).
In the present study the importance of calcium ions
and acetylcholine, as an element in the Ach/NO/cGMP signaling
pathway in the vascular contraction induced by ANG II and PHE
through metabotropic receptors (AT1 and α1-adrenergic,
respectively) was compared with the action of caffeine, a ryanodin
receptor agonist, in ER. Ji et al explained that the lack of
impact of acetylcholine and sodium nitroprusside on contraction
caused by caffeine is likely due to the fact that NO can
selectively block the release of calcium ions from the ER through
an IP3-dependent pathway (27). Perez-Zoghbi et al concluded
that NO, acting via the cGMP-PKG pathway, induced airway SMC
relaxation by predominately inhibiting the release of
Ca2+ via the IP3 receptor to decrease the frequency of
agonist-induced Ca2+ oscillations (26).
The action of ANG II and PHE was mediated by two
pools of calcium, and caffeine induced the contraction with the
parti cipation of calcium released from intracellular stores. The
relaxing effect of acetylcholine on responses stimulated by ANG II
and PHE indicates the participation of nitric oxide in modulating
the reactivity of arteries to the studied metabotropic receptor
agonists. No effect of acetylcholine on caffeine action suggests
that the pathway associated with nitric oxide does not interfere
with the vascular contraction induced by the ryanodin receptor.
References
|
1.
|
Karaki H, Ozaki H, Hori M, et al: Calcium
movements, distribution, and functions in smooth muscle. Pharmacol
Rev. 49:157–230. 1997.PubMed/NCBI
|
|
2.
|
Stewart K, Zhang Y and Davidge ST: Aging
increases PGHS-2-dependent vasoconstriction in rat mesenteric
arteries. Hypertension. 35:1242–1247. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Barton M, Cosentino F, Brandes RF, Moreau
P, Shaw S and Lüscher TF: Anatomic heterogeneity of vascular aging:
role of nitric oxide and endothelin. Hypertension. 30:817–824.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Cernadas MR, Sanchez de Miguel L and
Garcia-Duran M: Expression of constitutive and inducible nitric
oxide synthases in the vascular wall of young and aging rats. Circ
Res. 83:279–286. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Gasparo M, Catt KJ, Inagami T, Wright JW
and Unger TH: International union of pharmacology. XXIII. The
angiotensin II receptors. Pharmacol Rev. 52:415–472.
2000.PubMed/NCBI
|
|
6.
|
Mehta PK and Griendling KK: Angiotensin II
cell signaling: physiological and pathological effects in the
cardiovascular system. Am J Physiol Cell Physiol. 292:C82–C97.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Seasholtz TM, Gurdal H, Wang HY, Cai G,
Johnson MD and Friedman E: Heterologous desensitization of the rat
tail artery contraction and inositol phosphate accumulation after
in vitro exposure to phenylephrine is mediated by decreased levels
of Gαq and Gαi. J Pharmacol Exp Ther.
283:925–931. 1997.
|
|
8.
|
Drake MT, Shenoy SK and Lefkowitz RK:
Trafficking of G protein-coupled receptors. Circ Res. 99:570–582.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Keef KD, Hume JR and Zhong J: Regulation
of cardiac and smooth muscle Ca2+ channels
(CaV1.2a,b) by protein kinases. Am J Physiol Cell
Physiol. 281:C1743–C1756. 2001.PubMed/NCBI
|
|
10.
|
Breitwieser GE: G protein-coupled receptor
oligomerization: implications for G protein activation and cell
signaling. Circ Res. 94:17–27. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Nauli SM, Williams JM, Akopov SE, Zhang L
and Pearce WJ: Developmental changes in ryanodine- and
IP3-sensitive Ca2+ pools in ovine basilar artery. Am J
Physiol Cell Physiol. 281:C1785–C1796. 2001.PubMed/NCBI
|
|
12.
|
Valdés JA, Hidalgo J, Galaz JL, Puentes N,
Silva M, Jaimovich E and Carrasco MA: NF-B activation by
depolarization of skeletal muscle cells depends on ryanodine and
IP3 receptor-mediated calcium signals. Am J Physiol Cell Physiol.
292:C1960–C1970. 2007.PubMed/NCBI
|
|
13.
|
Somlyo AP and Somlyo AV: Ca2+
sensitivity of smooth muscle and non-muscle myosin II: modulated by
G proteins, kinases, and myosin phosphatase. Physiol Rev.
83:1325–1358. 2003.
|
|
14.
|
Grzesk G and Szadujkis-Szadurski L:
Modulation of the reactivity of α-adrenergic receptors by 8Br-cGMP
and angiotesin II. Ann Acad Med Bydg. 17:5–10. 2003.
|
|
15.
|
Grzesk G and Szadujkis-Szadurski L:
Pharmacometric analysis of α1-adrenoceptor function in pretreated
with lipopolysaccharides rat tail artery. Pol J Pharmacol.
53:605–613. 2001.
|
|
16.
|
Grzesk G, Trajder A, Szadujkis-Szadurska
K, Krzyzanowski M and Szadujkis-Szadurski L: Role of α-adrenergic
receptor reserve in different post-synaptic responses to exogenous
agonists in the bisected rat vas deferens. Prog Med Res. 3:13–16.
2005.
|
|
17.
|
Furchgott RF and Zawadzki JW: The
obligatory role of endothelial cells in relaxation of arterial
smooth muscle by acetylocholine. Nature. 288:373–376. 1980.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Murad F: The nitric oxide-cyclic GMP
signal transduction system for intracellular and intercellular
communication. Recent Prog Horm Res. 49:239–248. 1994.PubMed/NCBI
|
|
19.
|
Ignarro LJ: Endothelium-derived nitric
oxide: actions and properties. FASEB J. 3:31–36. 1989.PubMed/NCBI
|
|
20.
|
Xe L, Eu JP, Meissner G and Stamler JS:
Activation of the cardiac calcium release channel (ryanodine
receptor) by poly-S-nitrosylation. Science. 279:234–237. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Sun J, Xin C, Eu JP, Stamler JS and
Meissner G: Cysteine-3635 is responsible for skeletal muscle
ryanodine receptor modulation by NO. Proc Natl Acad Sci USA.
98:11158–11162. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Lim G, Venetucci L, Eisner DA and Casadei
B: Does nitric oxide modulate cardiac ryanodine receptor function?
Implications for excitation - contraction coupling. Cardiovasc Res.
77:256–264. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Moncada S, Palmer RM and Higgs EA: Nitric
oxide: physiology, pathophysiology, and pharmacology. Pharmacol
Rev. 43:109–142. 1991.
|
|
24.
|
Bender AT and Beavo JA: Cyclic nucleotide
phosphodiesterases: molecular regulation to clinical use. Pharmacol
Rev. 58:488–520. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Slupski M, Szadujkis-Szadurski L, Grzesk
G, et al: Guanylate cyclase activators influence reactivity of
human mesenteric superior arteries retrieved and preserved in the
same conditions as transplanted kidneys. Transplant Proc.
39:1350–1353. 2007. View Article : Google Scholar
|
|
26.
|
Perez-Zoghbi JF, Bai Y and Sanderson MJ:
Nitric oxide induces airway smooth muscle cell relaxation by
decreasing the frequency of agonist-induced Ca2+
oscillations. J Gen Physiol. 135:247–259. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Ji J, Benishin CG and Pang PKT: Nitric
oxide selectively inhibits intracellular Ca++ release
elicited by inositol trisphosphate but not caffeine in rat vascular
smooth muscle. J Pharmacol Exp Ther. 285:16–21. 1998.PubMed/NCBI
|
|
28.
|
Szadujkis-Szadurski R, Tafil-Klawe M,
Szadujkis-Szadurska K, et al: Effect of acetylocholine on reactions
induced by 2 contraction agents - angiotensin II and caffeine. Med
Biol Sci. 24:53–58. 2010.
|
|
29.
|
Szadujkis-Szadurska K, Szadujkis-Szadurski
R, Szadujkis-Szadurski L, et al: The influence of ischemia and
reperfusion injury on the reactivity of arteries induced by
angiotensin II and Bay K8644. Med Biol Sci. 24:47–52. 2010.
|
|
30.
|
Slupski M, Szadujkis-Szadurska K,
Szadujkis-Szadurski R, et al: Nitric oxide and thromboxane A2
modulate pulmonary pressure after ischemia and intestinal
reperfusion. Transplant Proc. 38:334–337. 2006. View Article : Google Scholar : PubMed/NCBI
|