Introduction
Cardiac hypertrophy has historically been considered
to be an adaptive response; however, prolonged hypertrophy is
associated with increased risk of sudden death or progression to
heart failure (1). Recent studies
have also demonstrated an antihypertrophic effect of κ-opioid
receptor activation in cardiac myocytes. For example, U50,488H, a
selective κ-opioid receptor agonist, inhibits the effects of
norepinephrine, an α-adrenoceptor agonist, on the electrically
induced intracellular Ca2+ transient in cardiac myocytes
(2). We also demonstrated that
U50,488H inhibits the Ca2+ transient and cardiac
hypertrophy induced by isoprenaline, a β-adrenoceptor agonist
(3). Emerging evidence indicates
that KATP activation reduces the remodeling process and
inhibits cardiac hypertrophy. For example, the KATP
opener nicorandil has been shown to reduce myocardial remodeling in
rats (4), whereas the putative
mitoKATP opener diazoxide inhibited phenylephrine
(PE)-induced cardiac hypertrophy in rat neonatal cardiomyocytes
(5). Thus, these studies indicate
a direct antihypertrophic effect of KATP activation in
the heart. Cardiac KATP channels are composed of SUR2A
and Kir6.2 subunits (6). There are
two types of KATP channels, namely the mitochondrial
KATP channel (mitoKATP) and the sarcolemmal
KATP channel (sarcKATP). Previous studies
have found that the opening of mitoKATP also plays an
important role in cardiac protection, such as in ischemic
preconditioning (7). The
mitoKATP channel has been established to play a critical
role in various types of preconditioning, whereas that of the
sarcKATP channel is controversial (8).
Although the mechanism of the antihypertrophic
effect of κ-opioid receptors is uncertain, we can refer to the
relationship of KATP channels and κ-opioid receptor in
ischemic preconditioning (IP). Previous studies have shown that the
opening of KATP channels and activation of κ-opioid
receptor exert cardio-protective effects against ischemic and
reperfusion (I/R) injury. In IP, U50,488H reduced the infarct size
induced by I/R in the rat and intracellular Ca2+
([Ca2+]i). The infarct-reducing effect of
U50,488H was reversed by blockade of the KATP channel,
which abolished the protective effect of preconditioning with
U50,488H (9). It has also been
shown that activation of PKC prevented the
[Ca2+]i overload and conferred
cardioprotection against hypoxic insults, and blockade of the
mitoKATP channel attenuated the effects of PKC
activation (10). κ-opioid
receptor signaling was impaired in cardiac hypertrophy due to a
defect in the coupling of PKC signaling with its effector (11). δ1-opioid receptor
mediates a potent cardioprotective effect via protein kinase C and
the mitochondrial KATP channel (12) and Wang et al (13) demonstrated that the mitochondrial
KATP channel is dependent on PKC for protection against
calcium and ischemia-induced injury.
In view of this body of evidence and the finding
that KATP opener and κ-opioid receptor agonist attenuate
hypertrophy, we hypothesized that the direct antihypertrophic
effects of κ-opioid receptor stimulation may involve
KATP activation and likely occur via the PKC pathway.
Accordingly, the present study was designed to determine whether
KATP channels mediate the antihypertrophic effect of
κ-opioid receptors in neonatal rat ventricular myocytes and, if so,
to assess and identify the nature of KATP involvement in
mediating the anti-hypertrophic effect of κ-opioid receptor
activation.
Materials and methods
Chemicals
Trans-(±)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)-cyclohexyl]-benzeneacetamid
methanesulfonate salt (U50,488H, U50) was used as a selective
κ-opioid receptor agonist (14,15),
and nor-binaltorphimine (NBI) was used as an antagonist (16–18).
Phenylephrine (PE), an α-adrenoceptor agonist, was used to induce
hypertrophy. 5-Hydroxydecanoic acid (5-HD) was used as a specific
blocker of the mitochondrial ATP-sensitive potassium channel.
Glibenclamide was used as a nonselective KATP channel
blocker. Chelerythrine was used as the protein kinase C inhibitor.
The concentrations of U50,488H (19–21),
PE, 5-HD, glibenclamide (22) and
chelerythrine (12) were based on
previous studies. All drugs were initially dissolved in distilled
water and subsequently diluted in culture medium, except for
glibenclamide and Fura-2/AM, which were dissolved in dimethyl
sulphoxide (DMSO). The final concentration of DMSO was <0.1%,
which itself had no effect.
U50,488H, NBI, 5-HD, glibenclamide, PE,
chelerythrine, Fura-2/AM, trypsin and DMEM were obtained from Sigma
Chemical Co. (St. Louis, MO, USA). Calf serum was obtained from Si
Ji Qing Chemical Co., Hangzhou, China.
Culture of neonatal rat ventricular
myocytes
In the experiment 65 neonatal rats were used, and
the protocols were approved by the Committee of Liaoning Medical
College for the Use of Experimental Animals for Research and
Teaching. Sprague-Dawley rats, 2–3 days old, were sacrificed, and
the heart was removed immediately. The ventricles were separated
from the atrium, trisected, and digested with trypsin (Sigma) in
0.8 mg/ml for 20 min at 37°C. Ventricular myocytes were cultured as
described previously (21). The
supernatant was removed following centrifugation and the pellet was
re-suspended in fetal bovine serum. The above steps were repeated
4–6 times until the ventricle was completely digested. The cell
suspension was diluted to 1×106/ml and placed in 24-well
tissue culture plates in humidified 5% CO2/95% air at
37°C for 48 h. The culture medium comprised 15% heat-inactivated
fetal bovine serum, 84% Dulbecco's modified Eagle's medium (DMEM)
and 1% penicillin-streptomycin, conditions shown to enhance the
growth of cultured ventricular myocytes. Bromodeoxyuridine (0.1 mM)
was added to prevent non-myocyte proliferation without toxicity to
myocytes (23). In experiments
involving treatment with PE, U50, NBI, 5-HD, glibenclamide or
chelerythrine, a low-serum (0.4%) DMEM was used. Myocardial cells
become ‘quiescent’ in low-serum medium and grow without
multiplication and/or proliferation (24).
Determination of cellular protein
content
Cells were cultured for 72 h with various treatments
(72 h was chosen as preliminary studies showed that the maximum
effects were obtained at that time). Dishes were washed rapidly
three times with Hank's solution, the cells were dissolved in 1%
sodium dodecylsulphate (SDS), and the protein content was measured
using the method described by Lowry et al (25).
Estimation of cell volume
The volume of ventricular myocytes was calculated
from measurement of cell diameter (26). The medium was aspirated and cells
were washed rapidly three times with D-Hank's solution. Cells were
then treated with 0.3 ml of 0.1% trypsin per well at 37°C for 10
min and the process was terminated with 10% fetal bovine serum (0.2
ml/ well). Digested cells were collected and measured using an
inverted microscope. For measurements, four or five fields were
randomly selected from 16 or 20 fields and photographed at high
power (magnification, x400), and 80 individual cell areas were
calculated using CIAS Daheng computer photograph analysis system
(China Da Heng Co., Beijing, P.R. China).
Incorporation of
[3H]leucine
[3H]leucine uptake was used as an index
of protein synthesis. The medium from myocardial cells grown in
24-well plates was aspirated and replaced with a medium contaning 1
Ci [3H]leucine. Drugs were added and incubation was
continued for 48 h. The medium was then aspirated and cells were
washed rapidly three times with cold Hank's solution. They were
then lysed by addition of l ml per well 1% SDS. Lysates were
collected and precipitated by the addition of 1 ml 5%
trichloroacetic acid and then applied to fiberglass GF/C filters.
After washing three times with 5 ml Hank's solution, filters were
dried and transferred to vials containing 4 ml scintillation fluid
and the radioactivity was determined using liquid scintillation
counting (27). The radioactivity,
which represented the [3H]leucine incorporated into
newly synthesized protein, was expressed as cpm per well.
Loading of cells with Fura-2/AM
Myocytes were cultured in wells, each with a
coverslip. The coverslips with myocytes were incubated with
Fura-2/AM (4 μM) in medium for 25 min. The unincorporated dye was
removed by washing twice with fresh medium. To allow the Fura-2/AM
in the cytosol to de-esterify, the loaded cells were maintained at
room temperature (24–26°C) for 60 min prior to the measurement of
[Ca2+]i.
Measurement of cytosolic calcium
transient
A spectrofluorometric method was used to measure the
cytosolic Ca2+ transient, using Fura-2/AM as the
Ca2+ indicator. After loading with Fura-2/AM, the
coverslips with myocytes were transferred to a superfusion chamber
on the stage of an inverted microscope, which was coupled to a TILL
imaging system (Munich, Germany), and superfused with Hank's
buffer. The emitted light was filtered at 510 nm. Fluorescence
signals at 340 nm (F340) and 380 nm (F380) were recorded on a
personal computer for data processing and analysis. Maximal
fluorescence for each coverslip was obtained after addition of the
Ca2+ ionophore ionomycin (20 μM). Ethylene glycol
tetraacetic acid (EGTA) was added to a final concentration of 20 mM
for the Ca2+-free condition. Cytosolic [Ca2+]
was calculated by the following formula:
[Ca2+]i = Kd x (Sf2/
Sb2) x (R340/380 -
Rmin)/(Rmax - R340/380) (28). Kd is the dissociation constant of
Fura-2/AM for Ca2+ and was assumed to be 225 nM at 37°C.
R340/380 is the ratio of corrected fluorescence signals.
Rmax is the ratio obtained following ionomycin
treatment. Rmin is the ratio of the corrected signals
obtained after EGTA treatment. Sf2 and Sb2 represent the
emission intensities at 380 nm excitation at saturation and under
Ca2+-free conditions, respectively.
Western blotting
Cells were washed once with ice-cold PBS containing
100 μM sodium orthovanadate and solubilized in the lysis buffer (50
mM Tris-HCl, 137 mM NaCl, 10% glycerol, 100 μM sodium
orthovanadate, 1 mM phenylmethylsulfonylfluoride, 10 μg/ml
aprotinin, 10 μg/ml leupeptin, and 1% Nonident P-40; pH 7.4). After
centrifugation at 12,000 x g for 20 min, the supernatant was
removed. Cells were dissolved in buffer containing 65 mM Tris-HCl
(pH 6.8), 3% SDS, 10% glycerol, and 6 mol urea. After measurement
of protein concentration (BCA kit, Pierce, Rockford, IL, USA),
β-mercaptoethanol and bromophenol blue were added to the buffer for
electrophoresis. Protein (60 μg) thus obtained (for Kir6.2) was
separated on 10% SDS-PAGE and transblotted to polyvinylidene
difluoride membranes (BioRad, Hercules, CA, USA). The blots were
incubated at 4°C overnight with antibodies and the resulting bands
were detected using enhanced chemiluminescence. Antibodies to
Kir6.2 at Thr-276 (1:1000 dilution; Santa Cruz) were used to detect
the activated form of the kinase. Intensities in the resulting
bands were quantified using CAMIAS008 image analysis system.
Statistical analysis
All data are expressed as the mean ± SEM. For the
effects of drugs at various concentrations, analysis of variance
(one-way ANOVA) was used to compare the control and treatment
groups. The post-LSD test was used to evaluate differences between
two groups. P<0.05 was considered to indicate statistical
significance.
Results
Effects of U50,488H, glibenclamide, 5-HD
or chelerythrine on PE-induced enhancement of spontaneous
[Ca2+]i transients
PE (10 μM) significantly increased the resting
[Ca2+]i (Fig.
1D) and reduced the peak amplitude (Fig. 1B) of spontaneous
[Ca2+]i transients (Fig. 1A). Both were abolished by 1 μM
U50,488H, which had no effect on normal cells. The effect of
U50,488H was abolished by 1 μM NBI, 50 μM glibenclamide, 100 μM
5-HD and 2 μM chelerythrine, each of which alone had no effect.
None of the treatments had any effect on the increased frequency of
spontaneous [Ca2+]i transients (Fig. 1C).
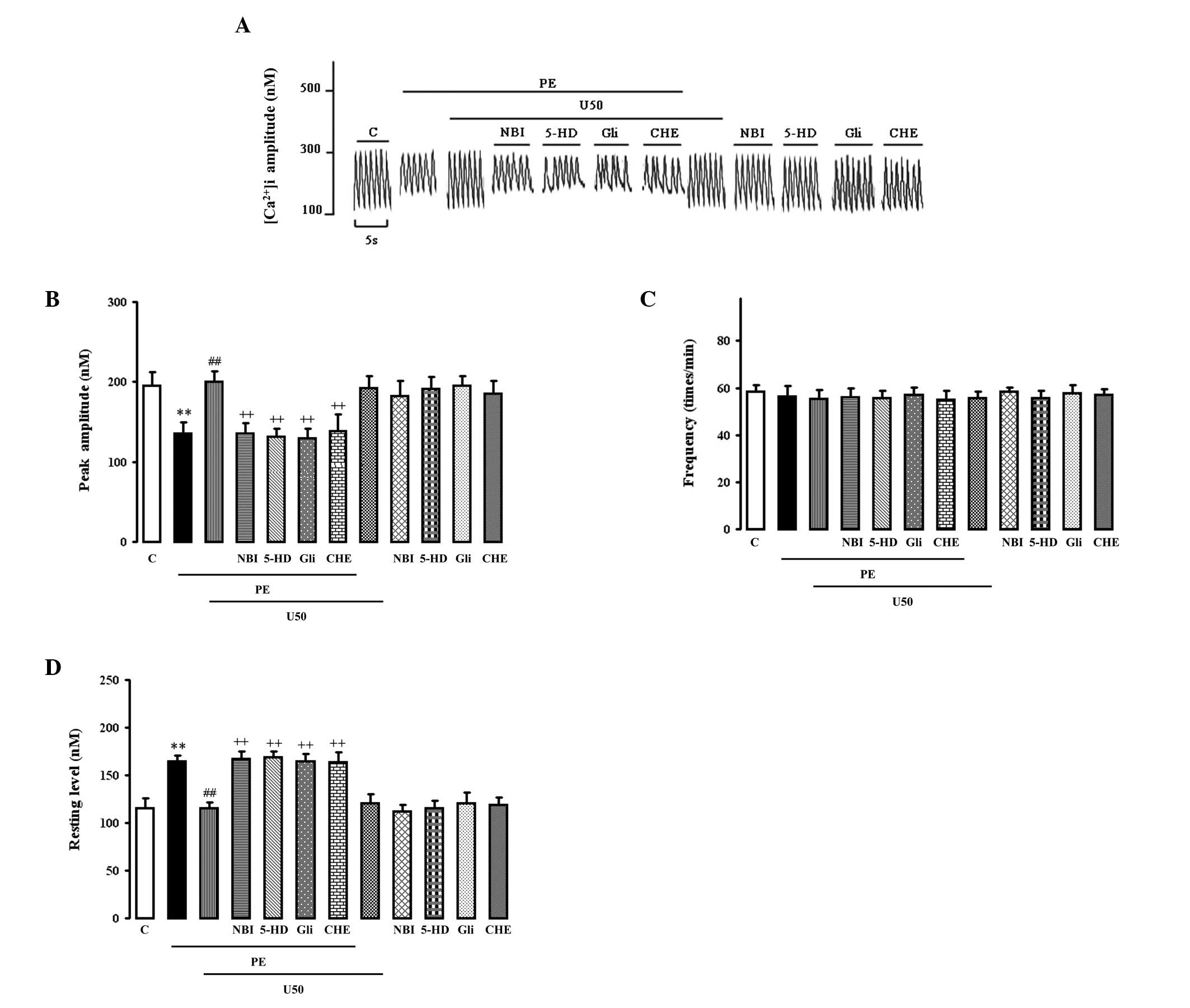 | Figure 1.Effects of U50,488H,
nor-binaltorphimine (NBI), glibenclamide (Gli), 5-hydroxydecanoic
acid (5-HD) or chelerythrine (CHE) on the (B) peak amplitude and
(C) frequency of the spontaneous [Ca2+]i
transient and (D) resting Ca2+ in cultured ventricular
myocytes from the neonatal rats treated with phenylephrine (PE).
(A) Representative tracings. The myocytes were cultured in wells
with a coverslip. After the cells were cultured for 2 days, the
coverslip with myocytes was incubated with Fura-2/AM at a
concentration of 4 μM in a medium for 25 min. The unincorporated
dye was removed by washing twice with fresh medium. Then the
cytosolic calcium transient of multiple cells were measured by the
TILL imaging system with a spec-trofluorometric method. The various
treatments were added respectively at certain time points. Values
are presented as mean ± SEM; n=4 in each group. **P<0.01 vs.
control; ##P<0.01 vs. PE group,
++P<0.01 vs. PE+U50 group. PE, 10 μM phenylephrine;
U50, 1 μM U50,488H; NBI, 1 μM nor-binaltorphi-mine; Gli, 50 μM
glibenclamide; 5-HD, 100 μM 5-hydroxydecanoic acid; CHE, 1 μM
chelerythrine. |
Effects of U50,488H, glibenclamide, 5-HD
or chelerythrine on PE-induced enhancement of total protein
content, cell size and [3H]leucine incorporation
PE (10 μM) significantly increased the total protein
content (Fig. 2A), cell size
(Fig. 2B) and
[3H]leucine incorporation (Fig. 2C) in myocytes. These effects were
abolished by 1 μM U50,488H, which itself had no effect. The
inhibitory effects of U50,488H were abolished by 1 μM NBI, 100 μM
5-HD, 50 μM glibenclamide and 2 μM chelerythrine, each of which
alone had no effect.
 | Figure 2.Effects of U50,488H,
nor-binaltorphimine (NBI), glibenclamide (Gli), 5-hydroxydecanoic
acid (5-HD) or chelerythrine (CHE) on (A) protein content, (B) cell
size and (C) [3H]leucine uptake in cultured ventricular
myocytes from the neonatal rats treated with phenylephrine (PE).
Methods and times of cell culture are as described in Materials and
methods. After the cells were cultured for 2-3 days, the medium was
changed to DMEM supplemented with 0.4% calf serum. The various
treatments were added to the medium as described in Materials and
methods and cultured for 48 h. Values are presented as mean ± SEM;
n=6. **P<0.01 vs. control; ##P<0.01 vs.
PE group; +P<0.05, ++P<0.01 vs. PE+U50
group. PE, 10 μM phenylephrine; U50, 1 μM U50,488H; NBI, 1 μM
nor-binaltorphimine; Gli, 50 μM glibenclamide; 5-HD, 100 μM
5-hydroxydecanoic acid; CHE, 1 μM chelerythrine. |
Effects of U50,488H, glibenclamide, 5-HD
or chelerythrine on Kir6.2 expression
U50,488H increased the expression of Kir6.2 in
myocytes exposed to PE, which itself had no effect (Fig. 3). Glibenclamide (50 μM), 5-HD (100
μM), NBI (1 μM) or chelerythrine (2 μM) abolished the effects of
U50,488H.
 | Figure 3.Effects of U50,488H,
nor-binaltorphimine (NBI), glibenclamide (Gli), 5-hydroxydecanoic
acid (5-HD) and chelerythrine (CHE) on Kir6.2 expression in
cultured myocardial cells from neonatal rats treated with
phenylephrine (PE). Apart from the cells (1×106 cells
per flask), the method and time of cell culture were the same as in
Fig. 2. The different treatments
were added to the medium at the same time and cultured for 48 h.
(A) Representative autoradiograms of Kir6.2 and β-actin. Lane 1,
normal control; lane 2, PE 10 μM; lane 3, PE 10 μM + U50 1 μM; lane
4, PE 10 μM + NBI 1 μM + U50 1 μM; lane 5, PE 10 μM + 5-HD 100 μM +
U50 1 μM; lane 6, PE 10 μM + Gli 50 μM + U50 1 μM; lane 7, PE 10 μM
+ CHE 1 μM + U50 1 μM. (B) Relative levels of Kir6.2 expressed as
the absorbance ratio of each group:control (%). Values are
presented as the mean ± SEM., n=4 in each group.
##P<0.01 vs. PE group. ++P<0.01 vs. PE
+ U50 group. PE, 10 μM phenylephrine; U50, 1 μM U50,488H; NBI, 1 μM
nor-binaltorphimine; Gli, 50 μM glibenclamide; 5-HD, 100 μM
5-hydroxydecanoic acid; CHE, 1 μM chelerythrine. |
Discussion
The present study demonstrated that administration
of U50,488H attenuated the increase in total protein content, cell
size and [3H]leucine incorporation induced by PE in rat
neonatal cardiomyocytes and that the effects were abolished by
nor-binaltorphimine. Having elucidated the identity of the
antihypertrophic effect of κ-opioid receptor, the goal of the study
focused on the signaling pathway involved. The initial hypothesis
was that the pathway was probably quite similar to that involved in
the κ-OR-mediated cardioprotective effect observed in previous
studies (9). The involvement of a
KATP channel in the adenosine receptor-mediated
antihypertrophic effect has also been well characterized (22). A previous study revealed that an
opioid agonist potentiated the opening of cardiac KATP
channels produced by a KATP channel opener to produce an
additive cardioprotective effect (29). To examine whether the same
mechanisms are at work in the present study, KATP
channel blockers were administered individually during treatment
with U50. Thus, our study demonstrated for the first time that the
antihypertrophic effect of κ-OR activation in rat neonatal
cardiomyocytes, at least with respect to PE-induced hypertrophy, is
dependent on KATP activation. This hypothesis is based
on the finding that the role of KATP in mediating the
antihypertrophic effect of κ-OR activation was clearly indicated by
the ability of pharmacological inhibitors of the channels to
abrogate the effect of U50. To determine the relative contributions
of mitochondrial KATP (mitoKATP) and
sarcolemmal KATP (sarcKATP) in the effect of
U50, we administered the nonspecific KATP blocker
glibenclamide or the mitoKATP specific blocker 5-HD,
both of which reversed the effect of U50. Surprisingly, the
reversing effect of the two blockers in response to U50 was
equivalent. This indicates that mitoKATP plays a
critical role. These data show that the KATP channel,
and most likely the mitochondrial channel, is a downstream effector
of the κ-opioid receptor.
The relative contributions of sarcolemmal and
mitochondrial KATP channel opening were revealed. The
consequences of sarc/mito KATP channel opening affect
various measures of antihypertrophy. SarcKATP channel
increases potassium efflux from the cell, hastening repolarization
and shortening the potential duration of the action. Mitochondrial
KATP channel activation is associated with numerous
effects, including membrane depolarization and changes in
Ca2+ homeostasis (30).
A brief depolarization of the mitochondrial membrane may exert
antihypertrophy by preventing Ca2+ entry into the
matrix. Therefore, it is likely that κ-OR activation opens
mitochondrial KATP and results in the decrease in the
mitochondrial membrane potential, thus reducing the driving force
of Ca2+ influx and attenuating the mitochondrial
Ca2+ overload induced by PE. Thus, a reduction in
'mitochondrial remodeling' may constitute a significant contributor
to the antihypertrophic effect of κ-OR. The study has also provided
the first evidence that the effect of the KATP channels
is accompanied by prevention/attenuation of the changes in
[Ca2+]i homeostasis, namely
[Ca2+]i overload, indicating that the
prevention/attenuation of the changes in
[Ca2+]i homeostasis may contribute, at least
partly, to the roles of the KATP channels by attenuation
of the [Ca2+]i overload in response to
PE-induced hypertrophy. PE significantly increased the resting
[Ca2+]i and reduced the peak amplitude and
spontaneous [Ca2+]i transients. Both were
abolished by U50,488H, which had no effect on normal cells. The
effect of U50,488H was abolished by 1 μM norbinaltorphimine,
glibenclamide, 5-HD and chelerythrine, each of which alone had no
effect.
Although our data support the hypothesis that κ-OR
activation inhibits PE-induced cardiac hypertrophy through the
opening of KATP, and particularly mitoKATP,
channels, via attenuation of the [Ca2+]i
overload in neonatal cardiomyocytes, the signaling pathway between
them remains uncertain. An additional set of experiments was
performed to determine whether PKC was involved in the signal
transduction pathway. A study by Seymour et al (31) showed that opioid receptors result
in activation of PKC, which then functions to open the
KATP channel to further enhance a cardioprotective
signal, and Wang et al (13) found that the mitochondrial KATP
channel is dependent on PKC for protection against calcium and
ischemic-induced injury. Opioid agonists act through Gi
protein-coupled opioid receptors, leading to the translocation and
activation of protein kinase C. Active PKC then initiates
cardioprotection through multiple kinase pathways, which
phosphorylate undetermined effectors (32,33).
Mitochondrial KATP channels opened by opioid-agonist
stimulation also play a critical role in PKC-mediated
cardioprotection (34,35). Therefore, we used chelerythrine, a
PKC inhibitor, to determine whether blocking PKC has any effect
upon the observed opioid receptor-mediated antihypertrophic effect
in myocytes. The data clearly show an inhibition of the effect of
U50, and indicate that PKC is involved in the pathway. Moreover, to
determine whether KATP channels act downstream of PKC we
assessed the expression of Kir6.2, a subunit of the KATP
channel, in the presence of chelerythrine. The data indicated that
U50 increased the expression of Kir6.2 in the presence of PE, which
suggests that U50 activated the opening of the KATP
channel. However, when chelerythrine was administered prior to U50,
the expression of Kir6.2 decreased compared with that of the PE+U50
group, which showed that the PKC inhibitor blocked the activation
of the KATP channel. This reveals that PKC acts upstream
of the KATP channel. Activation of G-protein-coupled
receptors may stimulate PKC to enhance KATP channel
activity.
In conclusion, our study shows an important role for
KATP in mediating the antihypertrophic effects of κ-OR
activation. Based on our results, we propose that the
antihypertrophic effect of κ-OR activation is dependent on
KATP channel activity, particularly mitoKATP
activity, via attenuation of [Ca2+]i
overload. Although PKC was associated with the antihypertrophic
effects of κ-OR receptor activation, the precise role of this
pathway and the role of PKC subtypes require further study.
Furthermore, evaluation of SUR2A and Kir6.2 mRNAs is clearly
warranted.
Acknowledgements
We thank Professor I.C. Bruce for the
advice, particularly regarding the language editing of the
manuscript, and Z.M. Qi, X.L. Xu and Z.H. Zong for their expert
technical assistance. This work was supported by the National
Natural Science Foundation (30973898/C190702).
References
|
1.
|
Frey N and Olson EN: Cardiac hypertrophy:
the good, the bad, and the ugly. Annu Rev Physiol. 65:45–79. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Yu XC, Li HY, Wang HX and Wong TM:
U50,488H inhibits effects of norepinephrine in rat
cardiomyocytes-cross-talk between kappa-opioid and beta-adrenergic
receptors. J Mol Cell Cardiol. 30:405–413. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Shan D, Wang HX, Su YH, Jing Y and Wong
TM: kappa-opioid receptor stimulation inhibits cardiac hypertrophy
induced by beta1-adrenoceptor stimulation in the rat. Eur J
Pharmacol. 555:100–105. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Sanada S, Node K, Asanuma H, Ogita H,
Takashima S and Minamino T: Opening of the adenosine
triphosphatesensitive potassium channel attenuates cardiac
remodeling induced by long-term inhibition of nitric oxide
synthesis: role of 70-kDa S6 kinase and extracellular
signal-regulated kinase. J Am Coll Cardiol. 40:991–997. 2002.
View Article : Google Scholar
|
|
5.
|
Xia Y, Rajapurohitam V, Cook MA and
Karmazyn M: Inhibition of phenylephrine induced hypertrophy in rat
neonatal cardiomyocytes by the mitochondrial KATP
channel opener diazoxide. J Mol Cell Cardiol. 37:1063–1067. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Seino S and Miki T: Physiological and
pathophysiological roles of ATP-sensitive K+ channels. Prog Biophys
Mol Biol. 81:133–176. 2003.
|
|
7.
|
Garlid KD, Paucek P, Yarov-Yarovoy V,
Murray HN, Darbenzio RB and D Alonzo AJ: Cardioprotective effect of
diazoxide and its interaction with mitochondrial ATP-sensitive K+
channels: possible mechanism of cardioprotection. Circ Res.
81:1072–1082. 1997.
|
|
8.
|
Oldenburg O, Cohen MV, Yellon DM and
Downey JM: Mitochondrial KATP channels: role in
cardioprotection. Cardiovasc Res. 55:429–437. 2002.
|
|
9.
|
Chen M, Zhou JJ, Kam KW, Qi JS, Yan WY and
Wong TM: Roles of KATP channels in delayed
cardioprotection and intracellular Ca2+ in the rat heart
as revealed by κ-opioid receptor stimulation with U50,488H. Br J
Pharmacol. 140:750–758. 2003.
|
|
10.
|
Light PE, Kanji HD, Fox JE and French RJ:
Distinct myoprotective roles of cardiac sarcolemmal and
mitochondrial KATP channels during metabolic inhibition
and recovery. FASEB J. 15:2586–2594. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Pei JM, Wang YM, Zhu YL, Chen M and Wong
TM: Signaling pathway mediated by kappa-opioid receptor is impaired
in cardiac hypertrophy. Acta Pharmacol Sin. 22:887–895.
2001.PubMed/NCBI
|
|
12.
|
Huh J, Gross GJ, Nagase H and Liang BT:
Protection of cardiac myocytes via delta(1)-opioid receptors,
protein kinase C, and mitochondrial KATP channels. Am J
Physiol Heart Circ Physiol. 280:H377–383. 2001.PubMed/NCBI
|
|
13.
|
Wang Y, Hirai K and Ashraf M: Activation
of mitochondrial ATP-sensitive K+ channel for cardiac protection
against ischemic injury is dependent on protein kinase C activity.
Circ Res. 85:731–734. 1999.
|
|
14.
|
Zukin RS, Eghbali M, Olive D, Unterwald EM
and Tempel A: Characterization and visualization of rat and guinea
pig brain kappa-opioid receptors: evidence for kappa 1 and kappa 2
opioid receptors. Proc Natl Acad Sci. 85:4061–4065. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Rothman RB, Bykov V, Costa BR, Jacobson
AE, Rice KC and Brady LS: Interaction of endogenous opioid peptides
and other drugs with four kappa opioid binding sites in guinea pig
brain. Peptides. 11:311–331. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Portoghese PS, Lipkowski AW and Takemori
AE: Binaltorphimine and nor-binaltorphimine, potent and selective
kappa-opioid receptor antagonists. Life Sci. 40:1287–1292. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Takemori AE, Ho BY, Naeseth JS and
Portoghese PS: Nor-binaltorphimine, a highly selective kappa-opioid
antagonist in analgesic and receptor binding assays. J Pharmacol
Exp Ther. 246:255–258. 1988.PubMed/NCBI
|
|
18.
|
Tortella FC, Echevarria E, Lipkowski AW,
Takemori AE, Portoghese PS and Holaday JW: Selective kappa
antagonist properties of nor-binaltorphimine in the rat MES seizure
model. Life Sci. 44:661–665. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Tai KK, Bian CF and Wong TM: kappa-opioid
receptor stimulation increases intracellular free calcium in
isolated rat ventricular myocytes. Life Sci. 51:909–913. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Ventura C, Spurgeon H, Lakatta EG,
Guarnieri C and Capogrossi MC: kappa and delta opioid receptor
stimulation affects cardiac myocyte function and Ca2+
release from an intra-cellular pool in myocytes and neurons. Circ
Res. 70:66–81. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Sheng JZ and Wong TM: Chronic U50,488H
abolishes inositol-1,4,5-trisphosphate and intracellular
Ca2+ elevations evoked by kappa-opioid receptor in rat
myocytes. Eur J Pharmacol. 307:323–329. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Xia Y, Javadov S, Gan TX, Pang T, Cook MA
and Karmazyn M: Distinct KATP channels mediate the
antihypertrophic effects of adenosine receptor activation in
neonatal rat ventricular myocytes. J Pharmacol Exp Ther. 320:14–21.
2007.
|
|
23.
|
Simpson P and Savion S: Differentiation of
rat myocytes in single cell cultures with and without proliferation
nonmyocardial cells. Cross-striations, ultrastructure, and
chromotropic response to isoproterenol. Circ Res. 50:101–116. 1982.
View Article : Google Scholar
|
|
24.
|
Berk BC, Vekshtein V, Gordon HM and Tsuda
T: Angiotensin II-stimulated protein synthesis in cultured vascular
smooth muscle cells. Hypertension. 13:305–314. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Lowry OH, Rosebrough NJ, Farr AL and
Randall RJ: Protein measurement with the Folin phenol reagent. J
Biol Chem. 193:265–275. 1951.PubMed/NCBI
|
|
26.
|
Zheng JS, Boluyt MO, Long X, O Neill L,
Lakatta EG and Crow MT: Extracellular ATP inhibits adrenergic
agonist-induced hypertrophy of neonatal cardiac myocytes. Circ Res.
78:525–535. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Luo JD, Xie F, Zhang WW, Ma XD, Guan JX
and Chen X: Simvastatin inhibits noradrenaline-induced hypertrophy
of cultured neonatal rat cardiomyocytes. Br J Pharmacol.
132:159–164. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Grynkiewicz G, Poenie M and Tsien RY: A
new generation of Ca2+ indicators with greatly improved
fluorescence properties. J Biol Chem. 260:3440–3450. 1985.
|
|
29.
|
Patel HH, Ludwig LM, Fryer RM, Hsu AK,
Warltier DC and Gross GJ: Delta opioid agonists and volatile
anesthetics facilitate cardioprotection via potentiation of
KATP channel opening. FASEB J. 16:1468–1470.
2002.PubMed/NCBI
|
|
30.
|
Holmuhamedov EL, Jovanovic S, Dzeja PP,
Jovanovic A and Terzic A: Mitochondrial ATP-sensitive K+ channels
modulate cardiac mitochondrial function. Am J Physiol.
275:1567–1576. 1998.
|
|
31.
|
Seymour EM, Wu SY, Kovach MA, Romano MA,
Traynor JR, Claycomb WC and Bolling SF: HL-1 myocytes exhibit PKC
and KATP channel-dependent delta opioid preconditioning.
J Surg Res. 114:187–194. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Fryer RM, Wang Y, Hsu AK and Gross GJ:
Essential activation of PKC-delta in opioid-initiated
cardioprotection. Am J Physiol Heart Circ Physiol. 280:H1346–1353.
2001.PubMed/NCBI
|
|
33.
|
Ping P, Zhang J, Pierce WM and Bolli R Jr:
Functional proteomic analysis of protein kinase C epsilon signaling
complexes in the normal heart and during cardioprotection. Circ
Res. 88:59–62. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Hu K, Duan D, Li GR and Nattel S: Protein
kinase C activates ATP-sensitive K+ current in human and rabbit
ventricular myocytes. Circ Res. 78:492–498. 1996.
|
|
35.
|
Fryer RM, Hsu AK, Eells JT, Nagase H and
Gross GJ: Opioid-induced second window of cardioprotection:
potential role of mitochondrial KATP channels. Circ Res.
84:846–851. 1999. View Article : Google Scholar : PubMed/NCBI
|














