Introduction
Melanoma is a major public health problem in many
countries. Effective treatment depends upon early diagnosis
followed by surgical excision with adequately wide margins. Medical
therapies have contributed little to enhancing the control of
established metastatic disease (1). Over the past 40 years no drug, or
combination of drugs, has shown any impact on the survival of
patients with metastatic melanoma; however, molecular pathways have
been recently identified that are central to melanoma growth and
apoptosis. These pathways are now under intense investigation as
potential therapeutic targets (1,2).
The cyclic nucleotide phosphodiesterase (PDE)
superfamily represents 11 gene families (PDE1-11) which differ in
their biochemical properties, regulation and sensitivity to
pharmacological agents. PDEs regulate signal transduction by
modulating intracellular levels of cyclic AMP (cAMP) and cGMP.
Three of the 11 PDE families (PDE4, PDE7 and PDE8) selectively
hydrolyze cAMP, 3 families (PDE5, PDE6 and PDE9) selectively
hydrolyze cGMP and 5 families (PDE1, PDE2, PDE3, PDE10 and PDE11)
hydrolyze both cyclic nucleotides (3).
PDE4 family members, which exclusively hydrolyze
cAMP, are widely expressed and play major regulatory roles. Much of
the functional knowledge of PDE4 enzymes was deduced from
experiments using highly selective inhibitors, targeted gene
knockout, small inhibitory RNA-mediated ablations or
dominant-negative mutations to disrupt endogenous intracellular
enzyme activities (4). Four genes
(PDE4A, PDE4B, PDE4C and PDE4D) encode more than 20 distinct PDE4
isoforms as a result of mRNA splicing and the use of distinct
promoters (5,6). The various isoforms encoded by a
single gene can differ in their enzymatic properties and tissue
distributions. Furthermore, these diverse isoforms each interact
with various distinct scaffolding proteins, leading to different
roles in compartmentalized intracellular signaling. The PDE4 family
has attracted considerable attention over the past decade as its
selective inhibitors have potential therapeutic use for a wide
range of major disease areas such as asthma, chronic obstructive
pulmonary disease, depression and Parkinson’s disease (4).
It is suggested that PDE4 is expressed in several
types of malignant tumor cells, where this family may induce
apoptosis (7) or regulate cell
migration (8). However, the
expression patterns and roles of PDE4 in malignant melanoma cells
are still unclear. In this study, we examined expression patterns
and roles of PDE4 in mouse B16-F10 melanoma cells.
Materials and methods
Cell culture
B16-F10 cells, a highly metastatic lung selected
cell subline derived from C57/BL6 murine melanoma, are frequently
used for the migration experiments (9). B16-F10 cells (American Type Culture
Collection, Manassas, VA, USA) were maintained in Dulbecco’s
modified Eagle’s medium (DMEM) containing 10% fetal bovine serum
(FBS) (Invitrogen Life Technologies, Carlsbad, CA, USA) at 37°C in
a humidified 5% CO2 atmosphere. The medium was changed 3
times/week.
Preparation of cell extracts for cAMP PDE
assay
B16-F10 cells were seeded at 2.5x105
cells/25 cm2 flask. After 3 days, the cells were washed
twice with phosphate buffered saline (PBS), harvested with a rubber
policeman and then homogenized in 1 ml of ice-cold homogenization
buffer (100 mM TES, pH 7.4, 10 μg/ml each of pepstatin,
leupeptin and aprotinin, 1 mM benzamidine, 0.5 mM Pefabloc, 1 mM
EDTA, 0.1 mM EGTA, 5 mM MgSO4 and 10% glycerol).
cAMP PDE assay
cAMP PDE activity was assayed as previously
described (10). Samples were
incubated at 30°C for 10 min in a total volume of 0.3 ml,
containing 50 mM HEPES, pH 7.4, 0.1 mM EGTA, 8.3 mM
MgCl2 and 0.1 μM [3H] cAMP (18,000
cpm). PDE4 activity was measured as the cAMP PDE activity inhibited
by 10 μM rolipram, a specific inhibitor of PDE4 (11).
RT-PCR
B16-F10 cells were seeded at 2.5x105
cells/25 cm2 flask. After 3 days, total RNA was isolated
from B16-F10 cells using the QuickGene RNA cultured cell kit S, and
from mouse heart using the QuickGene RNA tissue kit S (Fuji Photo
Film Co., Tokyo, Japan). Mouse kidney total RNA was purchased from
Stratagene (Santa Clara, CA, USA). First-strand cDNA was generated
from total RNA using a High Capacity RNA-to-cDNA™ kit
(Applied Biosystems, Foster City, CA, USA). PCR was performed with
specific oligonucleotide primer sets for PDE4A, PDE4B, PDE4C,
PDE4D, PDE4B2 and PDE4D5 (Table
I). The PCR reaction was carried out in a total volume of 50
μl containing 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 1.5 mM
MgCl2, 200 μM dNTPs, 2.5 units
HotStarTaq™ DNA Polymerase (Qiagen, Hilden, Germany) and
10 μM sense and antisense primers. HotStarTaq™
DNA Polymerase was activated by incubation of the reactions at 95°C
for 15 min. For PDE4A, PDE4B, PDE4C, PDE4D and PDE4B2, this
activation step was followed by 30 cycles of amplification (94°C
for 1 min, 62°C for 1 min and 72°C for 1 min) and 72°C for 10 min.
For PDE4D5, this was followed by 30 cycles of amplification (94°C
for 30 sec, 60°C for 30 sec and 72°C for 30 sec) and 72°C for 7
min. Products were subjected to electrophoresis on 2% agarose gels
and visualized by a SYBR-Green nucleic acid gel stain (Molecular
Probes, Inc., Eugene, OR, USA).
 | Table I.Primer sequences used for RT-PCR. |
Table I.
Primer sequences used for RT-PCR.
| Primer | Product size
(bp) |
|---|
| PDE4A |
5′-TTCAAGCTGCTGCAAGAAGA-3′
5′-TTCCTGAGGACCTGGATACG-3′ | 225 |
| PDE4B |
5′-GAACAAATGGGGCCTTAACA-3′
5′-TTGTCCAGGAGGAGAACACC-3′ | 609 |
| PDE4C |
5′-CATGCTCAACCGTGAGTTGT-3′
5′-TGGAACGTCTTGAGGAGGTC-3′ | 372 |
| PDE4D |
5′-GGAGCTTGTCACCTTCTTGG-3′
5′-GTGGGCTTTAAGTTGCTCCA-3′ | 181 |
| PDE4B2 |
5′-GAGCACCAGAAAGAGCTTGG-3′
5′-CAGACACCTGGTTCCCTGAT-3′ | 345 |
| PDE4D5 |
5′-ATCCGTTTCTCCCAAGCTCT-3′
5′-CAGGCTAGCCAAGACCTGAG-3′ | 365 |
Growth experiments
The cells were plated in a 96-well plate (100
cells/well) and allowed to adhere for 24 h. The cells were then
cultured in the absence or presence of different concentrations of
reagents for 6 days. MTS
[3-(4,5-dimethyl-thiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium,
inner salt] assays were performed using a CellTiter 96®
Aqueous One Solution cell proliferation assay (Promega, Madison,
WI, USA) and cell numbers were calculated.
cAMP concentration in mouse B16-F10
melanoma cells
B16-F10 cells were plated in a 6-well plate
(3x105 cells/well). After a 48-h incubation, the cells
were incubated with medium containing the indicated concentrations
of reagents for 30 min. Intracellular cAMP content was determined
using a Cyclic AMP EIA kit (Cayman Chemical Co., Inc., Ann Arbor,
MI, USA).
Migration assay
Cell migration was assayed as previously described
(12). Briefly, cells
(2.5x104 cells in DMEM medium containing 0.1% FBS with
the indicated concentrations of reagents) were transferred to
8-μm pore BD Falcon™ cell culture inserts (BD Biosciences,
Bedford, MA, USA). The inserts, which were coated with fibronectin
(6 μg/membrane) on the lower surface of the membrane, were
placed in companion wells containing DMEM medium supplemented with
20% FBS as a chemoattractant. Following a 12-h incubation, the
inserts were removed and the cells on the upper surface of the
membranes were wiped with a cotton swab. Cells on the lower surface
of the membrane were then fixed and stained with Diff-Quick Stain™
(Sysmex Co., Ltd., Kobe, Japan). The cells on the membrane were
then counted under a microscope.
Statistical analysis
Differences were statistically analyzed using the
Tukey-Kramer multiple comparisons test or Student’s t-test
(Fig. 5C). Significance was
defined as a calculated P-value <0.05.
Results
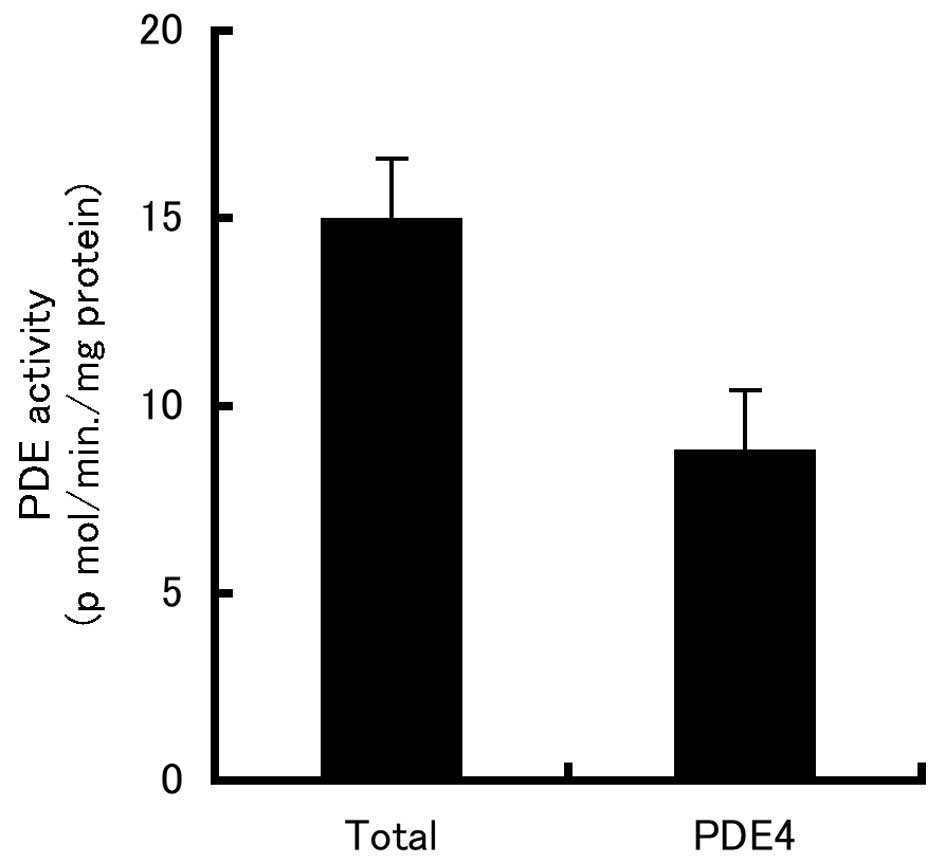
PDE4 activity in B16-F10 cells
To test whether PDE4 is expressed in B16-F10 cells,
we used the PDE4-specific inhibitor, rolipram. As shown in Fig. 1, ∼60% of the total PDE activity in
extracts of B16-F10 cells was PDE4 activity.
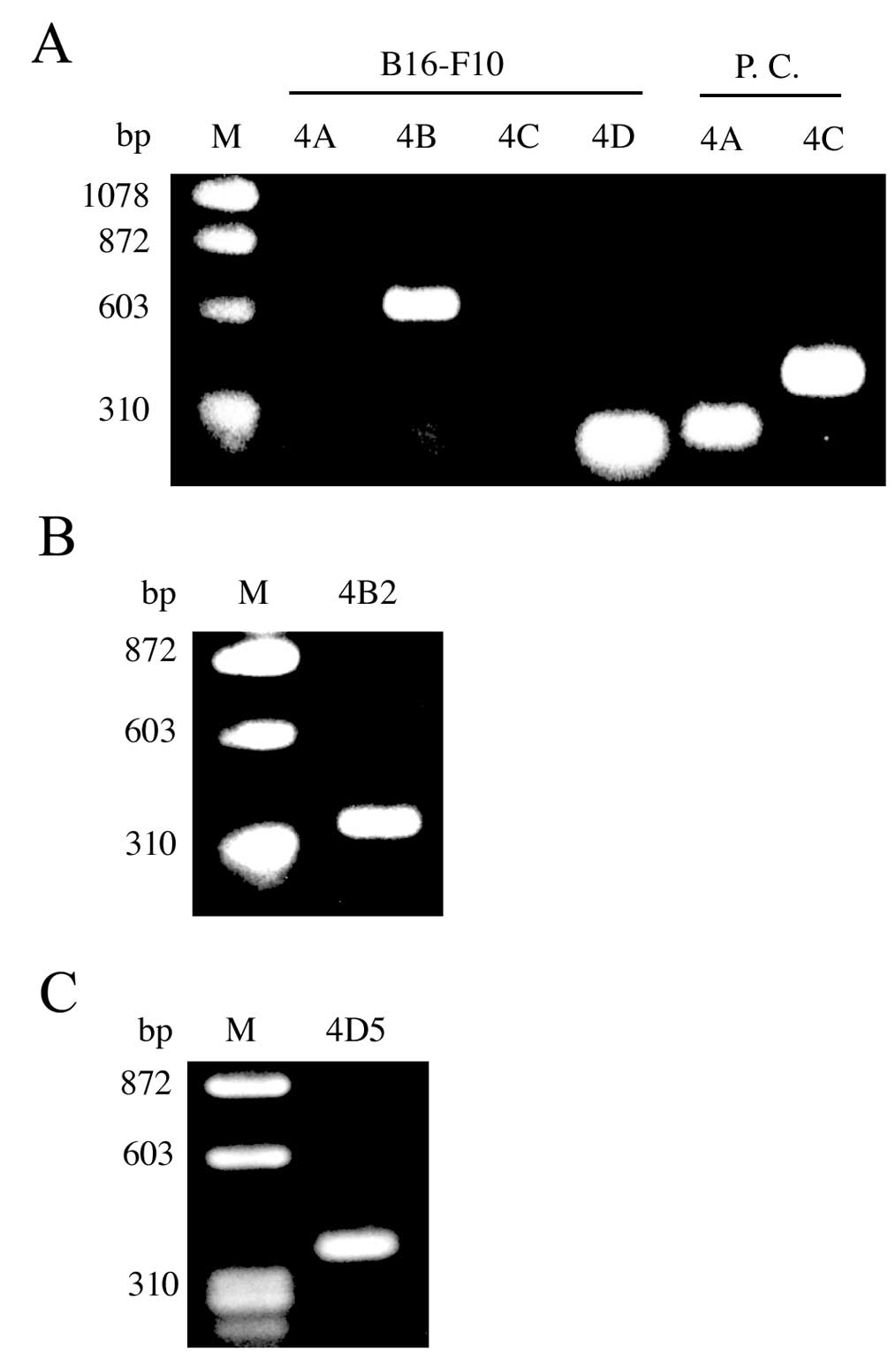
PDE4 mRNA expression in B16-F10
cells
To determine which PDE4 isoforms (PDE4A, PDE4B,
PDE4C and/or PDE4D) are expressed in B16-F10 cells, we performed
RT-PCR. PDE4B and PDE4D, but not PDE4A or PDE4C, mRNA were detected
by RT-PCR in total RNA from B16-F10 cells (Fig. 2A). Over 20 different PDE4 isoforms
are generated from 4 genes through the use of specific promoters
and alternative mRNA splicing. Interestingly, we were able to
detect PDE4B2 and PDE4D5 mRNA in the RNA from B16-F10 cells
(Fig. 2B and C).
Effect of a cAMP analog and PDE4-specific
inhibitors on B16-F10 cell growth
While the cAMP analog, 8-bromo-cAMP (8-Br-cAMP),
suppressed cell growth after 6 days of treatment (Fig. 3A), no effects were observed after
treatment with the PDE4-specific inhibitors rolipram and
denbufylline (Fig. 3B and C). To
verify the efficacy of rolipram and denbufylline, we next measured
cAMP concentration in the treated cells. After a 30-min treatment,
the intracellular cAMP concentration was significantly increased by
rolipram and denbufylline at concentrations of 10 and 50 μM
for both drugs (Fig. 3D and E).
These results confirm that rolipram and denbufylline suppress PDE4s
and increase intracellular cAMP concentration.
Effect of a cAMP analog and PDE4-specific
inhibitors on B16-F10 cell migration
We next investigated whether PDE4 is necessary for
B16-F10 cell migration. We found that 8-Br-cAMP inhibited B16-F10
cell migration (Fig. 4A).
Interestingly, the PDE4-specific inhibitors, rolipram and
denbufylline inhibited migration in a dose-dependent manner
(Fig. 4B and C). It was confirmed
that there were no significant changes in the number of cells after
having been cultured with each agent (data not shown).
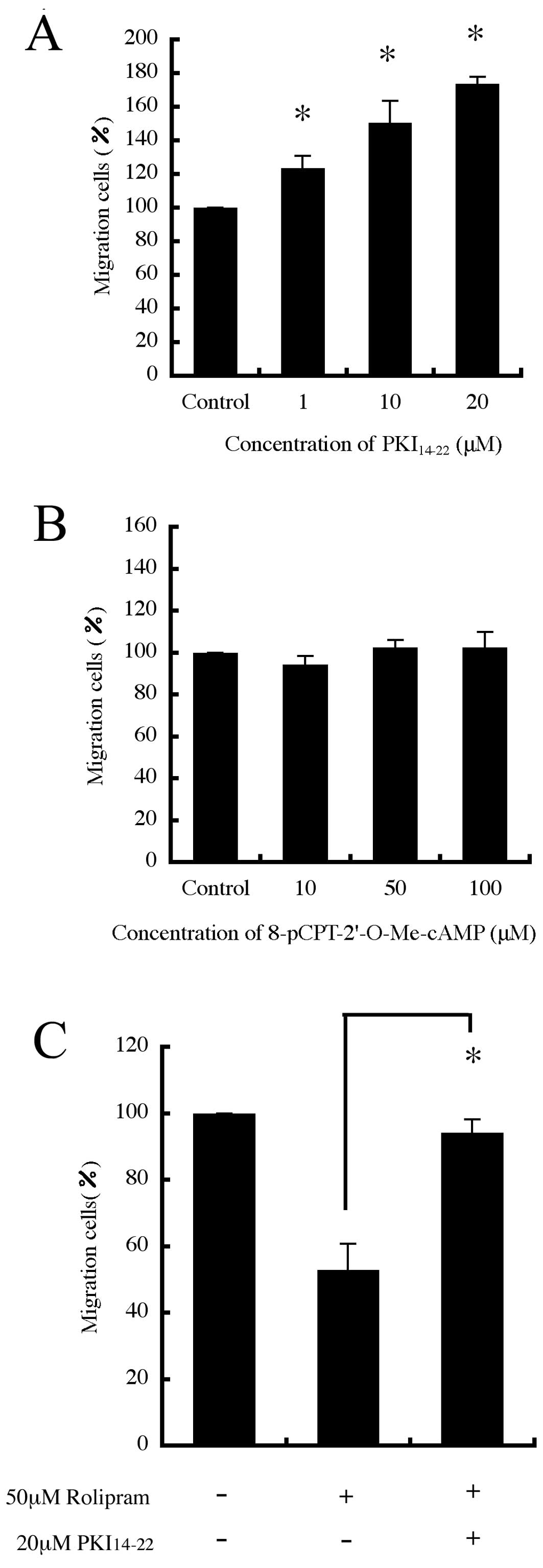
Effect of PKA and Epac on B16-F10 cell
migration
Since 8-Br-cAMP activates both protein kinase A
(PKA) and Epac (13), we decided
to investigate whether targeting these proteins affects cell
migration. We first used H-89, a widely used PKA inhibitor, and
found that H-89 promoted cell migration in a dose-dependent manner
(data not shown). However, H-89 inhibits other protein kinases at
concentrations similar to those affecting PKA (14). Therefore, we also studied the
effect of a PKA inhibitor, PKI14–22, and found that
PKI14–22 also promoted cell migration in a
dose-dependent manner (Fig. 5A).
In contrast, an Epac activator, 8-pCPT-2′-O-Me-cAMP that only
activates Epac without any concomitant activation of PKA on cell
migration, had no effect (Fig.
5B). Furthermore, the inhibitory effect of rolipram on cell
migration was reversed by PKI14–22 (Fig. 5C). It was confirmed that there were
no significant changes in the number of cells after having been
cultured with each agent (data not shown).
Discussion
Surgical resection of a malignant melanoma provides
an effective first-line therapy; however, melanoma can progress to
an aggressive, metastasizing form of the disease with extremely
poor patient survival rates (15).
Induction of apoptosis by increasing cAMP levels with PDE4
inhibitors has been demonstrated for colon cancer and chronic
lymphoid leukemia, suggesting that PDE4 inhibitors, in clinical
development for different therapeutic indications, could be a new
strategy for cancer treatment (16). Although previous studies report the
expression of PDEs in melanoma, their contribution to tumor
pathology remains obscure (17).
Previous studies have demonstrated that proliferation in B16-F10
cells (a highly metastatic lung selected subline derived from
murine melanoma) is suppressed and cell morphology is altered by
pentoxifylline, a well-known nonspecific PDE inhibitor with the
possible involvement of cAMP-PKA signaling (18). However, the study failed to
determine which PDEs were involved. Here, we found that the main
cAMP-PDE activity in extracts of B16-F10 cells involved PDE4, and
we detected both PDE4B and PDE4D mRNA by RT-PCR. PDE4 isoforms have
a role in achieving compartmentalized signaling in cells by
interacting with various scaffold proteins. PDE4B and PDE4D have
been reported to have distinct patterns of distribution in mouse
embryonic fibroblasts and control cAMP in different cellular
subdomains (19). These two
isoforms might be play different roles in B16-F10 cells. Over 20
different isoforms are generated from 4 genes through the use of
specific promoters and alternative mRNA splicing. Recently,
Marquette et al (16)
reported that PDE4B2 and PDE4D5 are expressed in human melanoma
cell lines, whereas mouse melanocytes express only PDE4B2 and not
PDE4D. We also detected PDE4B2 and PDE4D5 mRNA in B16-F10 cells.
These data suggest that PDE4D5 may be important in malignant
melanoma.
Although 8-Br-cAMP suppressed B16-F10 cell
proliferation, PDE4 specific inhibitors did not show any effects on
proliferation. The PDE4 inhibitors were functional as they yielded
an elevated cAMP concentration. Abusnina et al (20), reported that treatment with 10
μM rolipram for 24 h had no effect on B16-F10 cell
proliferation, which is consistent with our results. We also tested
the effect of rolipram on proliferation for 6 days, but no change
was observed. It was suggested that either PDEs may form an
enzymatic barrier to cAMP diffusion or, alternatively,
compartmentalized PDEs may act as a sink to localized pools of cAMP
(19). Therefore, it is probable
that other PDEs are involved in proliferation. In B16-F10 cells, in
addition to PDE4, PDE8 activity was detected (20) as cAMP-PDE, and by RT-PCR, we
detected PDE1A, PDE1B, PDE1C, PDE7A, PDE10A and PDE11A (data not
shown).
Notably, PDE4-specific inhibitors suppress B16-F10
cell migration. Recent research demonstrated that RhoA and Rac1
GTPases induce B16-F10 cell motility (21). PKA, which is a major effector of
cAMP, induces phosphorylation of RhoA and regulates its function in
several cell types (22). It is
also reported that PKA-induced phosphorylation of RhoA, as a
consequence of PDE4 inhibition, provides a probable mechanism for
the reduced RhoA-GTP levels and subsequent effects on peripheral
microspike formation noted in rat embryo fibroblast REF52 cells
(22). We also found that
PKI14–22 promoted cell migration. To investigate whether
the inhibitory effect of PDE4 inhibition on B16-F10 motility is
mediated by enhanced PKA activation, we also tested the effects of
PKI14–22 on rolipram-mediated inhibition of migration.
PKI14–22 reversed rolipram-mediated inhibition by almost
100%. Furthermore, we used another effector of cAMP, Epac, which
serves as GEFs for the small GTPases Rap1 and Rap2 (22). In contrast to the effects seen with
PKA inhibition, 8-pCPT-2′-O-Me-cAMP had no effect on migration.
This suggests that PDE4 inhibition suppresses B16-F10 cell
migration by inducing downstream PKA activation. Furthermore,
Serrels et al (8) showed
that PDE4D5 regulates chemically induced squamous cell carcinoma
cell polarity. However, in REF 52 cells, it is unclear which PDE4
isoform(s) regulates microspike formation (22). PDE4 isoform(s) that function to
regulate cell migration may differ in different cell types. More
studies are required to determine which isoform(s), regulate cell
migration in a wider panel of cell lines.
In conclusion, our results suggest that PDE4
regulates cell migration via cAMP-PKA signaling in B16-F10 cells
and that cAMP-PDE4 signaling may be a new therapeutic target for
melanoma.
Acknowledgements
This study was supported by a grant
from the Okasan-Kato Foundation to Y. Watanabe and a Grant-in-Aid
for Scientific Research (B) from the Japan Society for the
Promotion of Science (18390537) to T. Murata.
References
|
1.
|
Thompson JF, Scolyer RA and Kefford RF:
Cutaneous melanoma. Lancet. 36:687–701. 2005. View Article : Google Scholar
|
|
2.
|
Eggermont AM and Robert C: New drugs in
melanoma: it’s a whole new world. Eur J Cancer. 47:2150–2157.
2011.
|
|
3.
|
Beavo JA, Houslay MD and Francis SH:
Cyclic nucleotide phosphodiesterase superfamily. Cyclic Nucleotide
Phosphodiesterases In Health and Disease. Beavo JA, Francis SH and
Hously MD: CRC Press; New York: pp. 3–17. 2007
|
|
4.
|
Hously MD, Schafer P and Zhang KY: Keynote
review: phosphodiesterase-4 as a therapeutic target. Drug Discov
Today. 10:1503–1519. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Hously MD: Underpinning compartmentalized
cAMP signaling through targeted cAMP breakdown. Trends Biochem Sci.
35:91–100. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Bolger GB, Conti M and Houslay MD:
Cellular function of PDE4 enzymes. Cyclic Nucleotide
Phosphodiesterases In Health and Disease. Beavo JA, Francis SH and
Hously MD: CRC Press; New York: pp. 99–129. 2007
|
|
7.
|
Kim DH and Lerner A: Type 4 cyclic
adenosine monophosphate phosphodiesterase as a therapeutic target
in chronic lymphocytic leukemia. Blood. 92:2484–2494.
1998.PubMed/NCBI
|
|
8.
|
Serrels B, Sandilands E, Serrels A, et al:
A complex between FAK, RACK1, and PDE4D5 controls spreading
initiation and cancer cell polarity. Curr Biol. 20:1086–1092. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Fidler IJ: Selection of successive tumour
lines for metastasis. Nat New Biol. 242:148–149. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Murata T, Taira M and Manganiello VC:
Differential expression of cGMP-inhibited cyclic nucleotide
phosphodiesterases in human hepatoma cell lines. FEBS Lett.
390:29–33. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Houslay MD, Sullivan M and Bolger GB: The
multienzyme PDE4 cyclic adenosine monophosphate-specific
phosphodiesterase family: intracellular targeting, regulation, and
selective inhibition by compounds exerting anti-inflammatory and
antidepressant actions. Adv Pharmacol. 44:225–342. 1998. View Article : Google Scholar
|
|
12.
|
Moutasim KA, Nystrom ML and Thomas GJ:
Cell migration and invasion assays. Methods Mol Biol. 731:333–343.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Holz GG, Chepumy OG and Schwede F:
Epac-selective cAMP analogs: new tools with which to evaluate the
signal transduction properties of cAMP-regulated guanine nucleotide
exchange factors. Cell Signal. 20:10–20. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Davies SP, Reddy H, Caivano M and Cohen P:
Specificity and mechanism of action of some commonly used protein
kinase inhibitors. Biochem J. 351:95–105. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Houslay MD: Hard times for oncogenic
BRAF-expressing melanoma cells. Cancer Cell. 19:3–4. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Marquette A, André J, Bagot M, Bensussan A
and Dumaz N: ERK and PDE4 cooperate to induce RAF isoform switching
in melanoma. Nat Struct Mol Biol. 18:584–591. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Lau E and Ronai Z: RAF-isotype switching:
from B to C through PDE. Nat Struct Mol Biol. 18:517–518. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Dua P and Gude RP: Antiproliferative and
antiproteolytic activity of pentoxifylline in cultures of B16F10
melanoma cells. Cancer Chemother Pharmacol. 58:195–202. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Blackman BE, Horner K, Heidmann J, et al:
PDE4D and PDE4B function in distinct subcellular compartments in
mouse embryonic fibroblasts. J Biol Chem. 286:12590–12601. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Abusnina A, Keravis T, Yougbare I, Bronner
C and Lugnier C: Anti-proliferative effect of curcumin on melanoma
cells is mediated by PDE1A inhibition that regulates the epigenetic
integrator UHRF1. Mol Nutr Food Res. 55:1677–1689. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Dua P and Gude RP: Pentoxifylline impedes
migration in B16F10 melanoma by modulating Rho GTPase activity and
actin organisation. Eur J Cancer. 44:1587–1595. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Fleming YM, Frame MC and Houslay MD:
PDE4-regulated cAMP degradation controls the assembly of
integrin-dependent actin adhesion structures and REF52 cell
migration. J Cell Sci. 117:2377–2388. 2004. View Article : Google Scholar : PubMed/NCBI
|