Introduction
Hydrogen sulfide (H2S), a novel and
important gaseous transmitter, is endogenously generated in
mammalian tissues predominantly by two
pyridoxal-5′-phosphate-dependent enzymes, cystathionine β-synthase
(CBS) and cystathio-nine γ-lyase (CSE), using either L-cysteine or
L-homocysteine as a substrate (1,2).
Recently, Shibuya et al (3)
observed that 3-mercaptopyruvate sulfurtransferase (3MST) in
conjunction with cysteine (aspartate) aminotranferase (CAT), as the
third H2S-producing enzyme, contributes significantly to
generating H2S from L-cysteine in the presence of
α-ketoglutarate. The distribution of these enzymes is species- and
tissue-specific, and CBS and CSE are present in the liver (4). H2S has been observed to
demonstrate a wide range of physiological functions and is
important for several pathological conditions. For example,
H2S opens K+-ATP channels in vascular and
gastrointestinal smooth muscle cells, neurons and pancreatic β
cells, regulating vascular tone, intestinal contractility,
neurotransmission and insulin secretion (5–8).
H2S has also been recognized to be involved in the
inflammatory response (9).
Bacterial endotoxins such as lipopolysaccharide
(LPS) induce excessive activation and upregulation of vascular
K+-ATP channels and hypotension, and substantially
reduce vascular sensitivity to vasoconstrictive agents (10,11).
H2S has been proposed as a potential endogenous ligand
for K+-ATP channels to induce K+-ATP
channel-mediated vasorelaxation (12) in several vascular tissues,
suggesting that H2S might be involved in endotoxic
shock.
As one of the most important organs in the body, the
liver functions in biotransformation and metabolism. Due to its
central location in regulating metabolism and response to both
physiological and pathological exogenous stimuli, all forms of
liver disease are accompanied by a certain degree of inflammation.
Therefore, the liver is vulnerable to damage from internal and
external factors, including inflammatory mediators, drugs and
poisons (13,14). Numerous studies have investigated
the overall function and mechanism of this organ and in related
research, the role of hydrogen sulfide in hepatic
ischemia-reperfusion injury has been reported (15). However, the significance of
endogenous H2S (particularly the CBS-H2S synthesis) in
endotoxemia at the hepatic cell level is rarely discussed.
In the present study, we used the BRL rat hepatic
cell line to imitate hepatocytes in vivo. We hypothesized
that the endogenous CBS-H2S synthesis participates in
the pathophysiological regulation of apoptosis induced by LPS. To
investigate the role of the CBS-H2S synthesis in the
pathogenesis of hepatocyte injury, we added LPS to BRL cells and
observed changes in endogenous CBS expression, H2S
concentration in the culture supernatant and regulation of
apoptosis following administration of CBS inhibitor or CBS small
interfering RNA (siRNA).
Materials and methods
Materials
LPS (Escherichia coli 0111:B4) and
aminooxyacetic acid (AOAA) were obtained from Sigma-Aldrich (St.
Louis, MO, USA). Other chemicals and reagents were of analytical
grade.
Cell culture
The normal rat hepatic cell line BRL was obtained
from the Cell Bank of the Chinese Academy of Sciences, Shanghai,
China. BRL cells were cultured at 37°C in a humidified incubator
with 95% air and 5% CO2 in RPMI-1640 medium (Gibco,
Invitrogen, Carlsbad, CA, USA) supplemented with 10%
heat-inactivated fetal bovine serum (FBS; Hyclone, Invitrogen), 100
U/ml of penicillin and 100 μg/ml of streptomycin. Cultured cells
were used at 70–80% confluence.
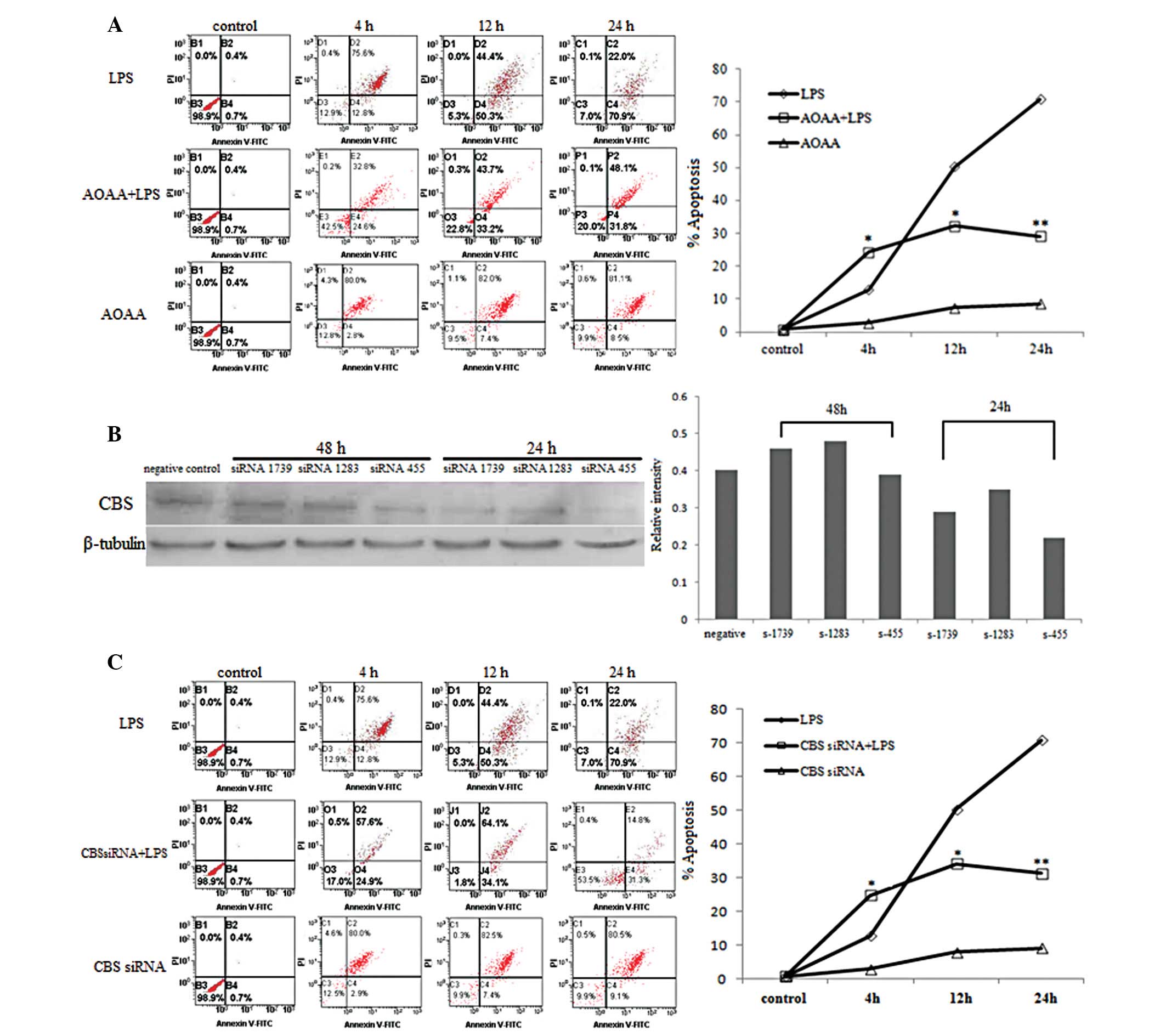
siRNA
CBS siRNA sequences were designed and synthetized by
Invitrogen: siRNA 455-s, 5′-CCAAGUGUGAGUUCUUCAATT-3′ and siRNA
455-a, 5′-UUGAAGAACUCACACU UGGTT-3′; siRNA 1283-s,
5′-CCAAGUUCUUGAGUGACAATT-3′ and siRNA 1283-a,
5′-UUGUCACUCAAGAACUUGGTT-3′; siRNA 1739-s,
5′-CCAUUGACCUGCUAAACUUTT-3′ and siRNA 1739-a,
5′-AAGUUUAGCAGGUCAAUGGTT-3′. One OD unit of each siRNA was
dissolved in 150 μl double-distilled H2O and 18 μl was
mixed with 6 μl Lipofectamine™ 2000 Transfection Reagent
(Invitrogen) in 550 μl serum-free medium for 30 min. This was added
to cells in 6-well plates. Western blot analysis was used to detect
the efficiency of gene silencing. The most efficient sequence was
used to transfect cells for flow cytometry (FCM) dectection.
CBS mRNA assay
Total RNA from BRL cells was extracted using TRIzol
reagent (Gibco, Invitrogen). Reverse transcription-polymerase chain
reaction (RT-PCR) was performed in a 0.2-ml tube containing 2 μl
tissue cDNA, 1 μl primer mixture of 5 μmol/l of each CBS-s,
5′-GAACCAGACGGAGCAAACAG-3′ and CBS-a, 5′-TGTAGAGGACTTTGCAGACT-3′
(Invitrogen), 1 μl of 2.5 mmol/l each dNTP, 1.5 μl of 1.5 mmol/l
MgCl2, 2.5 μl 10X PCR buffer and 1.25 U Taq DNA
polymerase, in 25 μl. After incubation at 95°C for 5 min, PCR was
performed at 94°C for 30 sec, 55°C for 30 sec and 72°C for 40 sec
for 30 cycles. The PCR products were separated on a 1.5% agarose
gel and stained with ethidium bromide. The optical density of the
band of CBS mRNA (572 bp) was measured using the Gel Documentation
System (Bio-Rad, Hercules, CA, USA). PCR products were amplified
again at 94°C for 30 sec, 55°C for 30 sec and 72°C for 30 sec for
20 cycles with rat GAPDH primers: GAPDH-s, 5′-C
CATGACAACTTTGGCATC-3′ and GAPDH-a, 5′-ATGTCA GATCCACAACGGA-3′
(Invitrogen). The optical density of the GAPDH mRNA band (262 bp)
was measured and the ratio of CBS mRNA/GAPDH mRNA was taken to be
the relative quantity of CBS mRNA.
Preparation of cell lysates for western
blot analysis
After treatment, BRL cells were homogenized in
ProteoJET™ mammalian cell lysis reagent supplemented with
ProteoBlock™ protease inhibitor cocktail (Fermentas, Amsterdam, The
Netherlands) and centrifuged at 4°C for 15 min at 16,000 × g. The
supernatants were collected and stored at −80°C. Protein
concentrations were determined by the Bio-Rad protein assay
(Bio-Rad).
Western blot analysis
The protein samples (30 μg) were separated by
SDS-polyacrylamide gel electrophoresis on 10% Tris-glycine
polyacrylamide gels and transferred to nitrocellulose membranes.
Nonspecific binding was blocked by incubation for 1 h in 5% nonfat
dry milk in PBST (0.05% Tween-20 in phosphate-buffered saline). The
blots were incubated overnight with primary antibody against CBS,
cytochrome c, or cleaved caspase-3 (Asp175; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) at 1:400 in 2.5% nonfat
dry milk in PBST, followed by washing 4 times with PBST and
incubating for 1 h with goat anti-rabbit horseradish
peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology,
Inc.) at 1:2,000 in 2.5% nonfat dry milk in PBST. The membranes
were washed and incubated in SuperSignal West Pico chemiluminescent
substrate (Pierce Chemical, Rockford, IL, USA) before exposure to
X-ray film (CL-XPosure; Pierce Chemical). The gels were calibrated
by protein Kaleidoscope standards (Bio-Rad). β-tubulin (Santa Cruz
Biotechnology, Inc.) was used as an internal control to normalize
for protein loading. Band intensity was quantified using LabWorks
Image Analysis software (UVP Upland, CA, USA).
Measurement of H2S
production
H2S production was measured as described
previously (12,16). Briefly, after treatment, cells were
collected and homogenized in 50 mM ice-cold potassium phosphate
buffer (pH 6.8). Flasks containing reaction mixture (100 mM
potassium phosphate buffer, 10 mM L-cysteine, 2 mM pyridoxal
5′-phosphate and 10% w/v cell homogenates) and center wells
containing 0.5 ml 1% zinc acetate and a piece of filter paper were
flushed with N2 and incubated at 37°C for 90 min. The
reaction was terminated by adding 0.5 ml 50% trichloroacetic acid
and flasks were incubated at 37°C for 60 min. The contents of the
center wells were transferred to test tubes each containing 3.5 ml
of water and 0.5 ml of 20 mM N, N-dimethyl-p-phenylenediamine
sulfate in 7.2 M HCl and 0.5 ml 30 mM FeCl3 in 1.2 M HCl
were added. The absorbance of the resulting solution at 670 nm was
measured after 20 min with a Multiskan® spectrum
microplate spectrophotometer (Thermo Scientific).
Apoptosis
The Annexin V-FITC/PI apoptosis detection kit was
purchased from Calbiochem (La Jolla, CA, USA). BRL cells were
dispersed by 0.25% trypsin and 1–5×105 cells were
collected and washed twice with phosphate buffer (pH 7.4). Cells
were suspended in 500 μl Annexin V binding buffer and mixed with 5
μl Annexin V binding buffer and 5 μl propidium iodide, and
incubated for 10 min in the dark at room temperature. Cell
apoptosis was detected by FCM (Cytomics FC500; Beckman-Coulter,
Miami, FL, USA; Ex=488 nm, Em=530 nm).
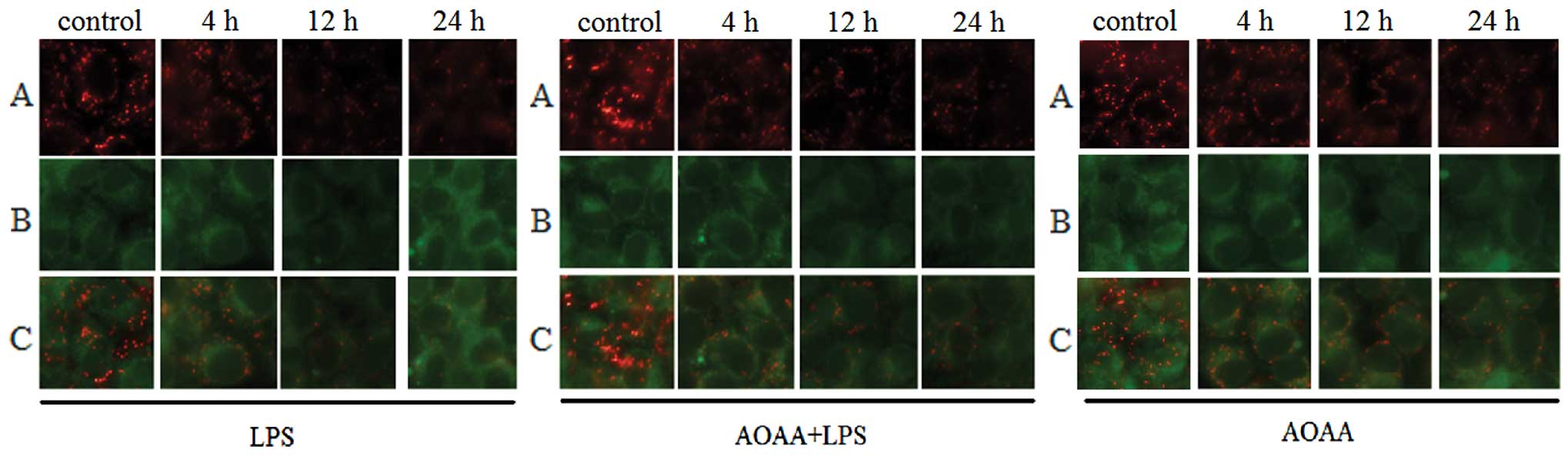
Mitochondrial membrane potential
BRL cells were cultured on coverslips prepositioned
in 6-well plates overnight. After treatment, cells were processed
with MitoCapture™ Apoptosis Detection kit (Calbiochem) and cultured
at 37°C in a humidified incubator with 95% air and 5%
CO2 for 20 min. MitoCapture is a cationic dye that
exists as a polymer in the mitochondria of normal cells and
produces a red fluorescence, or as a green fluorescent monomer in
the cytoplasm of apoptotic cells. Results were recorded by
fluorescence microscope (Eclipse 90i; Nikon) at x400
magnification.
Lactate dehydrogenase (LDH) release
assay
After treatment, BRL-cell medium was collected by
centrifugation at 850 × g for 3 min and stored at −70°C. Samples
were thawed and incubated at 37°C for 10 min. The tested sample
(200 μl) was added to each tube. The absorbance of the resulting
solution at 450 nm was measured with an automatic biochemical
analyzer (DXC600; Beckman-Coulter). LDH activity was calculated
against a sodium pyruvate calibration curve.
Statistical analysis
Results were expressed as mean ± SD. Comparison
between more than two groups was performed by one-way ANOVA and
Student Newman-Keuls test. P<0.05 was considered to indicate a
statistically significant result.
Results
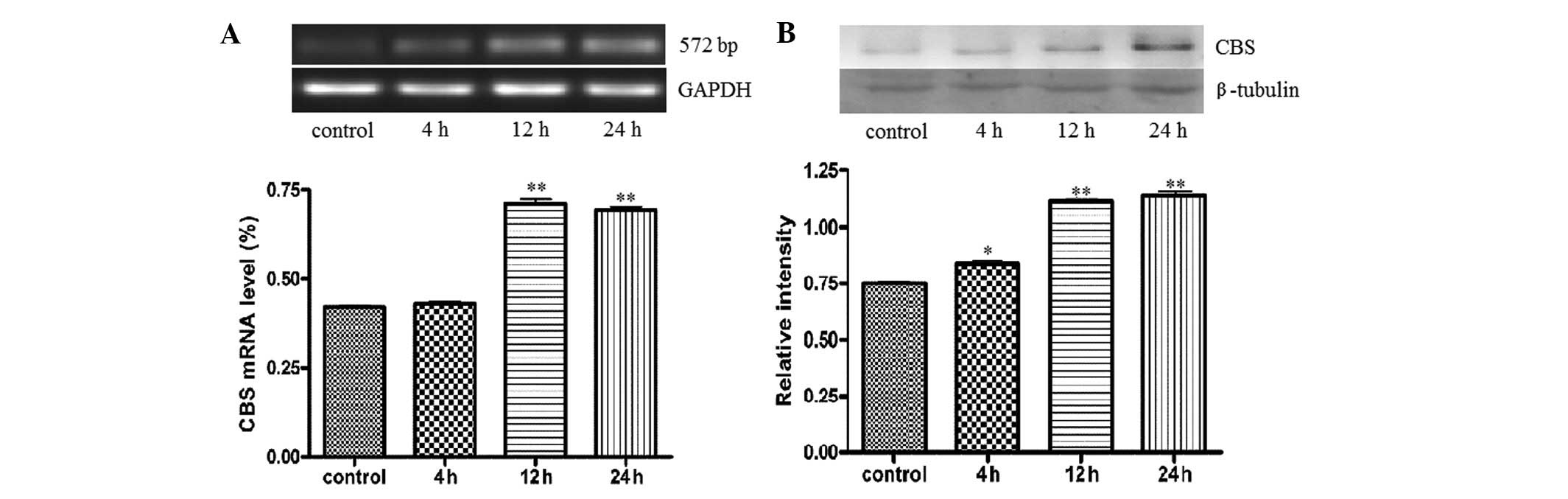
LPS treatment increases CBS and
H2S synthesis in BRL cells
The expression of CBS was measured in BRL cells
treated with 10 μg/ml LPS, the main ingredient in bacterial
endotoxin, which was used to injure BRL cells. RT-PCR demonstrated
that stimulation with LPS increased CBS mRNA over time from 4 to 24
h (P<0.05; Fig. 1A). Western
blot analysis showed that CBS protein in BRL cells was stimulated
by LPS, increasing significantly (P<0.05; Fig. 1B). The H2S production of
BRL cells increased markedly (P<0.01; Table I). We used LDH as a common index
for cell damage and observed that LDH in the culture medium
significantly increased from 4 to 24 h after LPS treatment
(P<0.05; Table II). In the
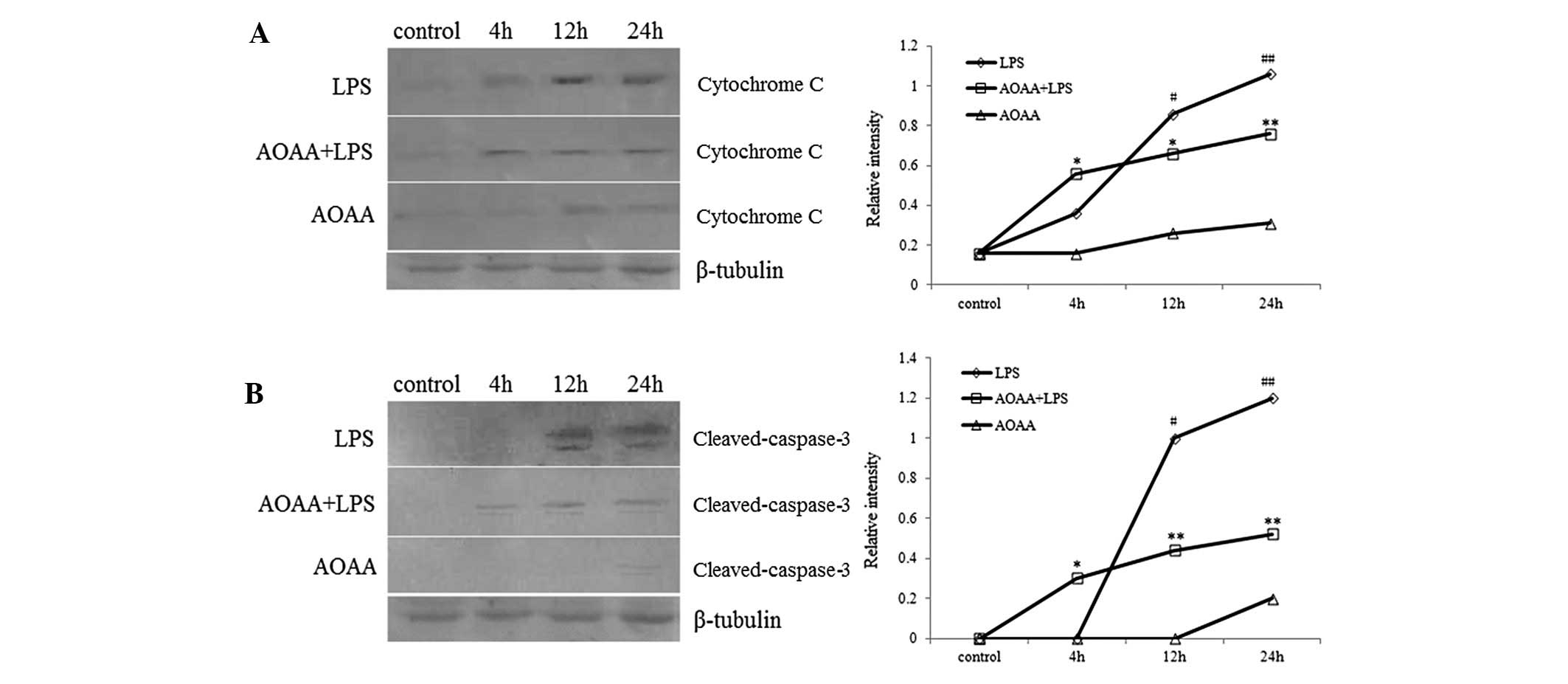
early stages of apoptosis, mitochondrial cytochrome c can be
detected in the cytoplasm. Western blot analyses for cytoplasmic
proteins indicated that cytochrome c appeared at 4 h and
markedly increased at 12 h after LPS treatment (P<0.05; Fig. 2A). Cleaved forms of caspase-3,
indicating the activated form, appeared at 12 h and increased at 24
h after LPS treatment (P<0.05; Fig.
2B). FCM detection showed that apoptosis of BRL cells increased
gradually from 4 to 24 h after LPS incubation (P<0.05; Table III and Fig. 3A). By contrast, variation in the
mitochondrial membrane potential decreased from 4 to 24 h
(P<0.05; Fig. 4A).
 | Table IAlteration of H2S
production in BRL cells (μmol/l). |
Table I
Alteration of H2S
production in BRL cells (μmol/l).
| Groups | Control | 4 h | 12 h | 24 h |
|---|
| LPS group | 26.19±4.93 | 36.23±4.23 | 46.73±3.44a | 58.23±2.52a |
| AOAA+LPS group | 26.17±4.57 | 24.33±2.43 | 19.25±2.24b | 16.98±4.02c |
| AOAA group | 26.18±4.45 | 24.74±3.98 | 18.66±4.31 | 13.42±2.45d |
| CBS siRNA+LPS
group | 26.17±4.77 | 24.26±3.23 | 19.77±2.27b | 16.22±3.55c |
| CBS siRNA
group | 26.19±4.66 | 24.01±4.11 | 18.46±3.37 | 13.13±3.01d |
| Table IIAlteration of culture supernatant LDH
(U/l). |
Table II
Alteration of culture supernatant LDH
(U/l).
| Groups | Control | 4 h | 12 h | 24 h |
|---|
| LPS group | 40.33±2.08 | 52.27±10.90 |
76.20±3.27a,b |
118.17±23.58b,c |
| AOAA+LPS group | 40.31±2.11 | 51.33±2.31 |
113.67±5.69c,d |
186.33±6.43c,d |
| AOAA group | 40.34±2.01 | 46.01±7.01 |
65.00±5.29a,b |
100.33±2.52c,d |
| CBS siRNA+LPS
group | 40.28±1.97 | 52.21±5.27 |
109.16±3.42c,d |
182.41±5.27c,d |
| CBS siRNA
group | 40.31±2.07 | 44.21±3.23 | 49.12±3.42 | 56.22±5.55 |
| Table IIIVariation of apoptosis rate in BRL
cells (%). |
Table III
Variation of apoptosis rate in BRL
cells (%).
| Groups | Control | 4 h | 12 h | 24 h |
|---|
| LPS group | 0.7±0.1 | 12.8±0.2a | 50.3±0.5b | 70.9±0.3b |
| AOAA+LPS group | 0.7±0.1 | 24.6±0.3c | 33.2±0.5c | 31.8±0.4d |
| AOAA group | 0.7±0.1 | 2.8±0.2 | 7.4±0.8 | 8.5±0.6 |
| CBS siRNA+LPS
group | 0.7±0.1 | 24.9±0.7c | 34.1±0.2c | 31.3±0.2d |
| CBS siRNA
group | 0.7±0.1 | 2.9±0.1 | 7.9±0.4 | 9.1±0.3 |
LPS-induced CBS-H2S synthesis
is inhibited by AOAA or CBS siRNA
BRL cells were pretreated with the CBS inhibitor
AOAA (3 mM) for 20 min prior to the addition of 10 μg/ml LPS. At 4,
12 and 24 h, production of H2S in the AOAA+LPS group
decreased significantly compared to the LPS alone group (P<0.05;
Table I) and LDH in the culture
supernatant of the AOAA+LPS group increased markedly at 12 h
(P<0.01; Table II). Western
blot analyses revealed that intracytoplasmic cytochrome c
clearly increased in the AOAA+LPS group at 4 h but decreased at 12
h after LPS treatment compared to the LPS alone group (P<0.05;
Fig. 2A). Although the level of
cleaved caspase-3 fragments in the AOAA+LPS group was higher than
in the LPS group at 4 h, LPS-induced detection of the cleaved
caspase-3 fragment was strongly attenuated by AOAA at both 12 and
24 h (P<0.01; Fig. 2B).
Detection by FCM indicated that apoptosis in the AOAA+LPS group
increased at 4 h and decreased at 12 and 24 h compared to the LPS
alone group (P<0.05; Table III
and Fig. 3A). The variation in
mitochondrial membrane potential in the AOAA+LPS group revealed a
trend opposite to apoptosis compared to the LPS group (Fig. 4).
After the transfection with CBS siRNA, the addition
of LPS to BRL cell cultures resulted in a significant decrease in
H2S production compared to the LPS alone group
(P<0.05; Table I). Apoptosis as
detected by FCM increased in the CBS siRNA+LPS group relative to
the LPS group at 4 h (P<0.05; Table
III and Fig. 3C). However,
apoptosis in the CBS siRNA+LPS group decreased significantly at 12
and 24 h compared to the LPS group (P<0.05; Table III and Fig. 3C).
The effect of AOAA on BRL cells
When the CBS inhibitor AOAA was added to BRL cells
at 3 mM, synthesis of H2S decreased over time
(P<0.05; Table I). A similar
affect was observed with transfection of BRL cells with CBS siRNA;
CBS downregulation markedly decreased the level of H2S
in BRL cells (P<0.05; Table I).
LDH in the culture supernatant of BRL cells increased in the
presence of AOAA (P<0.05; Table
II), but western blot analyses showed that AOAA treatment did
not alter the levels of intracytoplasmic cytochrome c or
cleaved caspase-3 (Fig. 2),
suggesting that AOAA had a minimal affect on BRL cell apoptosis.
AOAA had little effect on apoptosis or MMP. These results
demonstrate that LPS-induced apoptosis of BRL cells could be
blocked by the CBS inhibitor AOAA.
Discussion
H2S, named the third gaseous transmitter
following nitric oxide and carbon monoxide, may be trans-membrane
transported in a receptor-independent manner and activate various
cellular targets (17).
Particularly in the treatment of inflammation and
ischemia-reperfusion injury, the enzyme CSE and H2S are
an attractive pharmacological agent (9,18,19).
As the primary H2S-generating organ in vivo,
liver possesses the enzymes CSE and CBS (4). However, the enzyme CSE has been given
more attention for its involvement in physiological and
pathological conditions (20–24).
Whether the CBS-H2S synthesis plays an important role in
the modulation of hepatocyte apoptosis remains unknown.
In the present study, we treated the hepatic cell
line BRL with LPS to generate an acute injury model of hepatocytes
with the aim of observing the effect of the endogenous
CBS-H2S synthesis on inflammatory lesions of
hepatocytes. Our results indicate that CBS exists in the rat
hepatic cell line BRL, as previously described (2). LPS treatment results in the
upregulation of CBS mRNA and protein in BRL cells, with a
corresponding increase in total H2S production. The
apoptosis of BRL cells detected by FCM increased over time with a
corresponding decrease in MMP. The appearance of cytochrome
c in the cytoplasm increased and caspase-3 was activated by
LPS treatment. The addition of the CBS inhibitor AOAA or
transfection with CBS siRNA prior to LPS treatment in BRL cells
resulted in an increase in cell apoptosis but no significant change
in total H2S production at 4 h compared to an LPS alone
control. However, the apoptosis of BRL cells decreased at 12 h and
the H2S production also decreased, suggesting that
endogenous CBS has short-term anti-apoptosis effects, and promotes
apoptosis later. A possible explanation is CSE, another main
endogenous enzyme involved in H2S generation (22,23).
We hypothesize that since AOAA or CBS siRNA does not inhibit the
function of endogenous CSE, H2S synthesis continued, and
no significant change in total H2S production was
observed between the AOAA+LPS and LPS alone groups. However, over
time, endogenous CSE could not continue to enhance H2S
synthesis. A similar tendency was observed with cytochrome c
and cleaved caspase-3. Initially, cytoplasmic cytochrome c
was clearly enhanced in BRL cells pretreated with AOAA prior to the
addition of LPS, however, the expression then decreased compared to
cells treated with LPS alone. At 4 h, cleaved caspase-3 appeared,
suggesting activation, although the amount of cleaved caspase-3 was
less than that in cells stimulated by LPS alone after 12 h.
In conclusion, our results indicate that endogenous
CBS-H2S synthesis in BRL cells may regulate apoptosis
induced by LPS, partly by involving the mitochondrial pathway. The
course of regulation is complex and may be anti-apoptotic in the
short-term but pro-apoptotic in the long-term. These results may
provide references for the research and development of clinical
treatments. The specific mechanism of the regulation and
interaction of endogenous H2S synthesis requires further
investigation.
Acknowledgements
This study was supported by a project
funded by the Priority Academic Program Development of Jiangsu
Higher Education Institutions (PAPD).
References
|
1
|
Stipanuk MH and Beck PW: Characterization
of the enzymic capacity for cysteine desulphhydration in liver and
kidney of the rat. Biochem J. 206:267–277. 1982.PubMed/NCBI
|
|
2
|
Swaroop M, Bradley K, Ohura T, Tahara T,
Roper MD, Rosenberg LE and Kraus JP: Rat cystathionine-β-synthase.
Gene organization and alternative splicing. J Biol Chem.
267:11455–11461. 1992.
|
|
3
|
Shibuya N, Tanaka M, Yoshida M, Ogasawara
Y, Togawa T, Ishii K and Kimura H: 3-Mercaptopyruvate
sulfurtransferase produces hydrogen sulfide and bound sulfane
sulfur in the brain. Antioxid Redox Signal. 11:703–714. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhao W, Ndisang JF and Wang R: Modulation
of endogenous production of H2S in rat tissues. Can J
Physiol Pharmacol. 81:848–853. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang R: Two’s company, three’s a crowd:
can H2S be the third endogenous gaseous transmitter?
FASEB J. 16:1792–1798. 2002.
|
|
6
|
Moore PK, Bhatia M and Moochhala S:
Hydrogen sulfide: from the smell of the past to the mediator of the
future? Trends Pharmacol Sci. 24:609–611. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kimura Y, Dargusch R, Schubert D and
Kimura H: Hydrogen sulfide protects HT22 neuronal cells from
oxidative stress. Antioxid Redox Signal. 8:661–670. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang GD, Yang W, Wu LY and Wang R:
H2S, endoplasmic reticulum stress, and apoptosis of
insulin-secreting beta cells. J Biol Chem. 282:16567–16576.
2007.
|
|
9
|
Li L, Bhatia M and Moore PK: Hydrogen
sulphide - a novel mediator of inflammation? Curr Opin Pharmacol.
6:125–129. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gardiner SM, Kemp PA, March JE and Bennett
T: Regional haemodynamic responses to infusion of
lipopolysaccharide in conscious rat: effects of pre- or
post-treatment with glibenclamide. Br J Pharmacol. 128:1772–1778.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shi W, Cui N, Wu Z, Yang Y, Zhang S, Gai
HY, Zhu DL and Jiang C: Lipopolysaccharides up-regulate
Kir6.1/SUR2B channel expression and enhance vascular KATP channel
activity via NF-κB-dependent signaling. J Biol Chem. 285:3021–3029.
2010.PubMed/NCBI
|
|
12
|
Zhao W, Zhang J, Lu Y and Wang R: The
vasorelaxant effect of H2S as a novel endogenous gaseous
KATP channel opener. EMBO J. 20:6008–6016. 2001.PubMed/NCBI
|
|
13
|
Szabo G, Mandrekar P and Dolganiuc A:
Innate immune response and hepatic inflammation. Semin Liver Dis.
27:339–350. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Szabo G, Romics L Jr and Frendl G: Liver
in sepsis and systemic inflammatory response syndrome. Clin Liver
Dis. 6:1045–1066. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jha S, Calvert JW, Duranski MR,
Ramachandran A and Lefer DJ: Hydrogen sulfide attenuates hepatic
ischemia-reperfusion injury: role of antioxidant and antiapoptotic
signaling. Am J Physiol Heart Circ Physiol. 295:H801–H806. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang G, Sun X and Wang R: Hydrogen
sulfide-induced apoptosis of human aorta smooth muscle cells via
the activation of mitogen-activated protein kinases and caspase-3.
FASEB J. 18:1782–1784. 2004.PubMed/NCBI
|
|
17
|
Wang R: The gasotransmitter role of
hydrogen sulfide. Antioxid Redox Signal. 5:493–501. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li L, Bhatia M, Zhu YZ, Zhu YC, Ramnath
RD, Wang ZJ, Anuar FB, Whiteman M, Salto-Tellez M and Moore PK:
Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced
inflammation in the mouse. FASEB J. 19:1196–1198. 2005.PubMed/NCBI
|
|
19
|
Hui Y, Du J, Tang C, Bin G and Jiang H:
Changes in arterial hydrogen sulfide (H2S) content during septic
shock and endotoxin shock in rats. J Infect. 47:155–160. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang H, Zhi L, Moore PK and Bhatia M:
Role of hydrogen sulfide in cecal ligation and puncture-induced
sepsis in the mouse. Am J Physiol Lung Cell Mol Physiol.
290:1193–1201. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Collin M, Anuar FB, Murch O, Bhatia M,
Moore PK and Thiemermann C: Inhibition of endogenous hydrogen
sulfide formation reduces the organ injury caused by endotoxemia.
Br J Pharmacol. 146:498–505. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li L, Salto-Tellez M, Tan CH, Whiteman M
and Moore PK: GYY4137, a novel hydrogen sulfide-releasing molecule,
protects against endotoxic shock in the rat. Free Radic Biol Med.
47:103–113. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Awata S, Nakayama K, Suzuki I and Kodama
H: Effect of cysteine on the inactivation of cystathionine gamma
lyase by DL-propargylglycine. Acta Med Okayama. 43:329–335.
1989.PubMed/NCBI
|
|
24
|
Dulak NC and Shing YW: Large scale
purification and further characterization of a rat liver cell
conditioned medium multiplication stimulating activity. J Cell
Physiol. 90:127–137. 1977. View Article : Google Scholar
|