Introduction
Human telomerase is a specialized DNA polymerase
which controls the replication of chromosomal ends, or telomeres.
The majority of malignant tumors express telomerase but most normal
cells do not (1,2). Therefore, telomerase may be a good
candidate for targeted cancer gene therapy. Human telomerase
consists of 3 major components: the RNA component (hTER); the
telomerase-associated protein (hTEP1); and the telomerase catalytic
unit or human telomerase reverse transcriptase (hTERT) (3–5).
Both hTER and hTERT are necessary for telomerase activity, although
telomerase expression is predominantly regulated at the
transcriptional level of hTERT. Additionally, hTERT expression is
specific to human tumor cells, whereas hTER is present in normal
and tumor cells. Thus, the hTERT promoter has been extensively used
in targeted cancer gene therapy (6–13).
Vesicular stomatitis virus (VSV) is a
negative-stranded RNA rhabdovirus with a single molecule genome.
VSV includes 5 major proteins [nucleoprotein (N); phosphoprotein
(P); matrixprotein (M); glycoprotein (G) and polymerase (L)] and
selectively replicates in interferon (IFN)-resistant tumor cells.
VSV also induces host cell apoptosis via signaling through the
double-stranded RNA-dependent serine/threo-nine protein kinases,
Fas and Daxx (14–16). Studies have confirmed that VSV
efficiently suppresses the growth of syngeneic tumors in
immunocompetent mice and human tumor xenografts in nude mice and
prolongs the survival time of tumor-bearing animals. However,
severe adverse effects, including flu-like symptoms, encephalitis,
ventriculitis, oral vesicles and cervical lymphadenopathy, limit
the clinical applications of replication-competent VSV (17–20).
Matrix protein (MP) is important in viral assembly and
cytopathogenesis. Expression of MP alone causes a number of the
same cellular effects as infection with VSV via the inhibition of
host gene transcription and nucleocytoplasmic transport of host
RNAs and proteins (21–24). Previously, we constructed the
plasmid pVAX-M (VSV MP is under the control of the CMV promoter)
and revealed that pVAX-M alone or combined with radiation or DDP
efficiently inhibits solid tumor growth and significantly prolongs
survival (25–29). These findings suggest that VSV MP
is a promising agent for the treatment of tumors.
In the present study, in order to realize the
targeted anti-tumor effect of VSV MP, the plasmid phTERT-M (VSV MP
controlled by the hTERT promoter) was constructed. Subsequently, we
found that phTERT-M suppressed the tumor growth of the A549 model,
limited VSV MP overexpression to the tumor tissues and reduced VSV
MP expression in other organs more effectively than pVAX-M. These
results demonstrate that phTERT-M gene therapy is a more specific
and safer approach for the treament of human lung adenocarcinoma
than pVAX-M gene therapy.
Materials and methods
Cell line
The human lung adenocarcinoma A549 cell line was
obtained from the American Type Culture Collection (ATCC,
Rockville, MD, USA). It was cultured in RPMI-1640 medium
supplemented with 10% heat-inactivated fetal bovine serum (FBS),
100 U/ml of penicillin, 100 mg/ml of streptomycin and maintained in
a 37°C incubator with a humidified 5% CO2
atmosphere.
Plasmid construction, preparation of
cationic liposome and liposome-DNA complex
pVAX-M from the pVAX plasmid (Invitrogen Life
Technologies, San Diego, CA, USA) expressing wild-type VSV MP, was
constructed in our laboratory previously (25–29).
As a control, pure pVAX plasmid without VSV MP-cDNA was used as an
empty vector (null).
A 271-bp fragment containing the core promoter
region essential for the transactivation of hTERT was synthesized
and subcloned into the BglI/HindIII-digested pVAX-MP
plasmid to produce the phTERT-M plasmid (29,30).
The recombinant plasmid, phTERT-M, was confirmed to contain the
correct sequence by nucleotide sequencing.
The procedure for preparing plasmid and liposome was
performed as described in our previous studies (25–29).
DNA-liposome mixtures were prepared 30 min prior to use. DNA and
stock liposome were diluted in 5% dextrose in water (D5W) or
RPMI-1640 medium without serum and mixed in equal volumes, with a
DNA/liposome ratio of 1:3 (μg/μg). All reagents were diluted and
mixed at room temperature.
Cell viability assay in vitro
The cytotoxicity of phTERT-M-liposome mixtures or
pVAX-M-liposome mixtures on A549 cells was determined using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT;
Sigma, St. Louis, MO, USA) colorimetric assay. Cells (∼5,000
cells/100 μl medium) were seeded in each well of a 96-well plate
and incubated overnight. The cells were then treated with NS,
Lip-null (0.2 μg pVAX/0.6 μg liposome mixtures), Lip-phTERT-M (0.2
μg phTERT-M/0.6 μg liposome mixtures), Lip-pVAX-M (0.2 μg
pVAX-M/0.6 μg liposome mixtures) and etoposide (0.1 μg/ml),
respectively. Six wells were included in each group. After a 48-h
incubation, the medium was aspirated and 20 μl of 5 mg/ml MTT was
added per well and incubated at 37°C for 4 h; then supernatant
fluid was removed and 150 μl dimethyl sulfoxide (DMSO) was added
per well. Spectrometric absorbance at 540 nm was measured using a
microplate reader. The cell survival rate was assessed as percent
cell viability in terms of non-treated control cells.
Assessment of apoptosis in vitro
Cell apoptosis was evaluated by flow cytometry and
TUNEL assay (DeadEnd Fluorometric TUNEL System; Promega
Corporation, Madison, WI, USA). Flow cytometric analysis was
performed as previously reported (26,27).
Cells (∼2x105 cells/well) were plated in 6-well plates
and treated with NS, Lip-null (2 μg pVAX/6 μg liposome mixtures),
Lip-phTERT-M (2 μg phTERTP-M/6 μg lipo-some mixtures), Lip-pVAX-M
(2 μg pVAX-M/6 μg liposome mixtures) or etoposide (0.1 μg/ml).
After a 48-h incubation, the cells were collected and resuspended
in 1 ml hypotonic fluorochrome solution containing 50 μg/ml
propidium iodide (PI) in 0.1% sodium citrate with 0.1% Triton X-100
and then analyzed by flow cytometry. Cells appearing in the sub-G1
stage were considered as apoptotic cells. To quantify apoptotic
cells within the total tumor cells in vitro, TUNEL assays
were performed according to the manufacturer’s instructions. Cell
nuclei with dark green fluorescent staining were defined as
TUNEL-positive nuclei. TUNEL-positive nuclei were monitored by
fluorescence microscope (Leica, Bensheim, Germany). Five
equal-sized fields at x200 magnification were randomly chosen and
analyzed. The apoptotic index (AI) was defined as follows: AI (%) =
100 x (apoptotic cells/total tumor cells).
Animal studies
Female athymic BALB/c nude mice (SPF grade; 6- to
8-weeks old), were purchased from the Laboratory Animal Center of
Sichuan University and allowed to acclimate for 1 week before use.
All the animal studies were carried out in accordance with
institutional guidelines referring to animal use and care.
A549 cells (∼5×105) were injected into
the right flank of each nude mouse via subcutaneous inoculation.
When the size of the tumors reached ∼30 mm3, mice were
randomly assigned into 5 groups and treated with NS (100 μl),
Lip-null (10 μg pVAX/30 μg liposome mixtures, 100 μl), Lip-phTERT-M
(10 μg pVAX-MP/30 μg liposome mixtures, 100 μl), Lip-pVAX-M (10 μg
pVAX-M/30 μg liposome mixtures, 100 μl) or etopo-side (2 mg/kg).
The animals received 10 4-weekly intravenous administrations and
were monitored every 3 days for tumor burden, cachexia and other
abnormalities. Tumor sizes were measured using the formula A x
B2 x 0.52 (A, length; B, width; all in mm). All data are
presented as mean ± SD. Mice were sacrificed by cervical
dislocation when the volume of the tumor exceeded 6,000
mm3. Tissues of interest, including heart, liver,
spleen, lung, kidney and tumor, were excised and fixed in 10%
neutral-buffered formalin solution or frozen at −80°C.
Histological analysis
To analyze the targeted antitumor effect of phTERT-M
against the A549 model, immunohistochemical staining was performed
as described previously (26). The
tissues of each group were embedded in paraffin and cut into 3- to
5-μm sections. These sections were then deparaffinized in xylol and
rehydrated through a graded alcohol series. Antigen retrieval was
performed by autoclaving sections in 10 mM EDTA (pH 6.0) and
incubating them with rabbit immunoserum at 1:50 dilution, followed
by an incubation with biotinylated rat anti-rabbit antibody and
then streptavidin biotin reagents.
To quantify apoptotic cells within the tumor
sections, TUNEL assays (DeadEnd Fluorometric TUNEL System; Promega
Corporation) were performed according to the manufacturer’s
instructions. Five equal-sized fields at x200 magnification were
randomly chosen and analyzed. The apoptotic index (AI) was defined
as explained above.
Toxicity observation
No significant differences in weight were found
among the 5 groups. No adverse consequences in other gross
measures, including ruffling of fur, behavior, feeding and toxic
death, were observed in the Lip-phTERT-M group. Furthermore, no
significant differences in liver, lung, kidney, spleen, heart,
pancreas or brain were observed by hematoxylin and eosin
histological examination between the Lip-phTERT-M and NS
groups.
Statistical analysis
All the data were analyzed by the statistical
software SPSS 16.0. Data were assessed by ANOVA and Student’s
t-test. P<0.05 was considered to indicate a statistically
significant difference.
Results
The antitumor efficacy of Lip-phTERT-M on
A549 cells in vitro
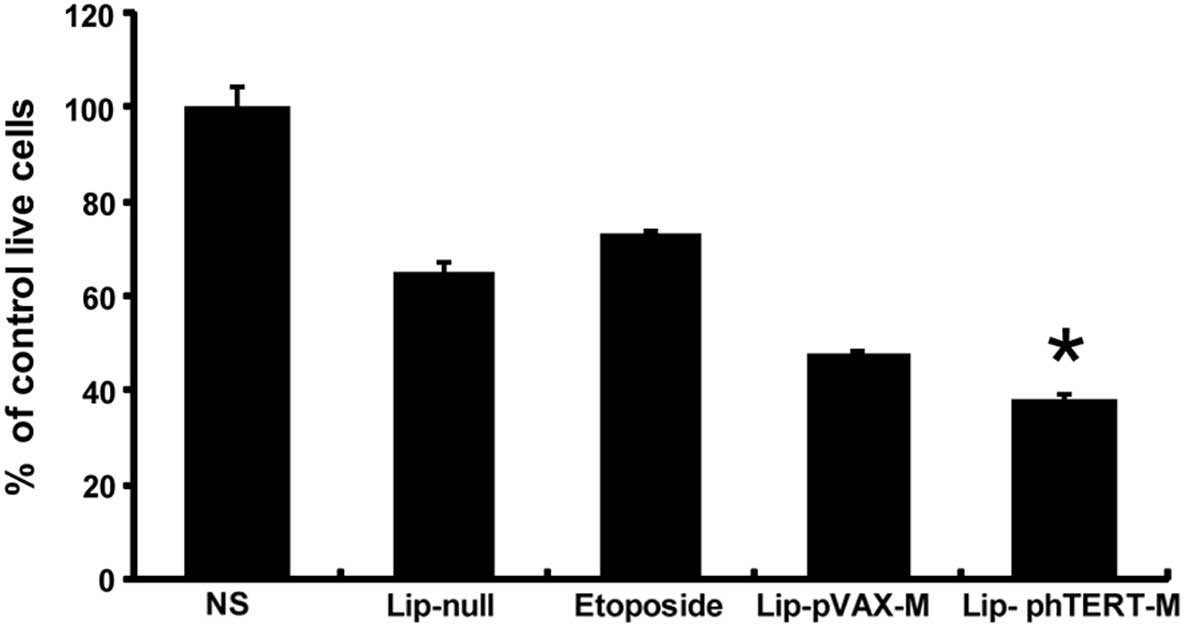
In order to evaluate the antitumor activity of
Lip-phTERT-M on A549 cells in vitro, cell viability assays
using MTT were performed. The results demonstrated that
Lip-phTERT-M reduced A549 cell growth more effectively than the
other groups (Fig. 1). The
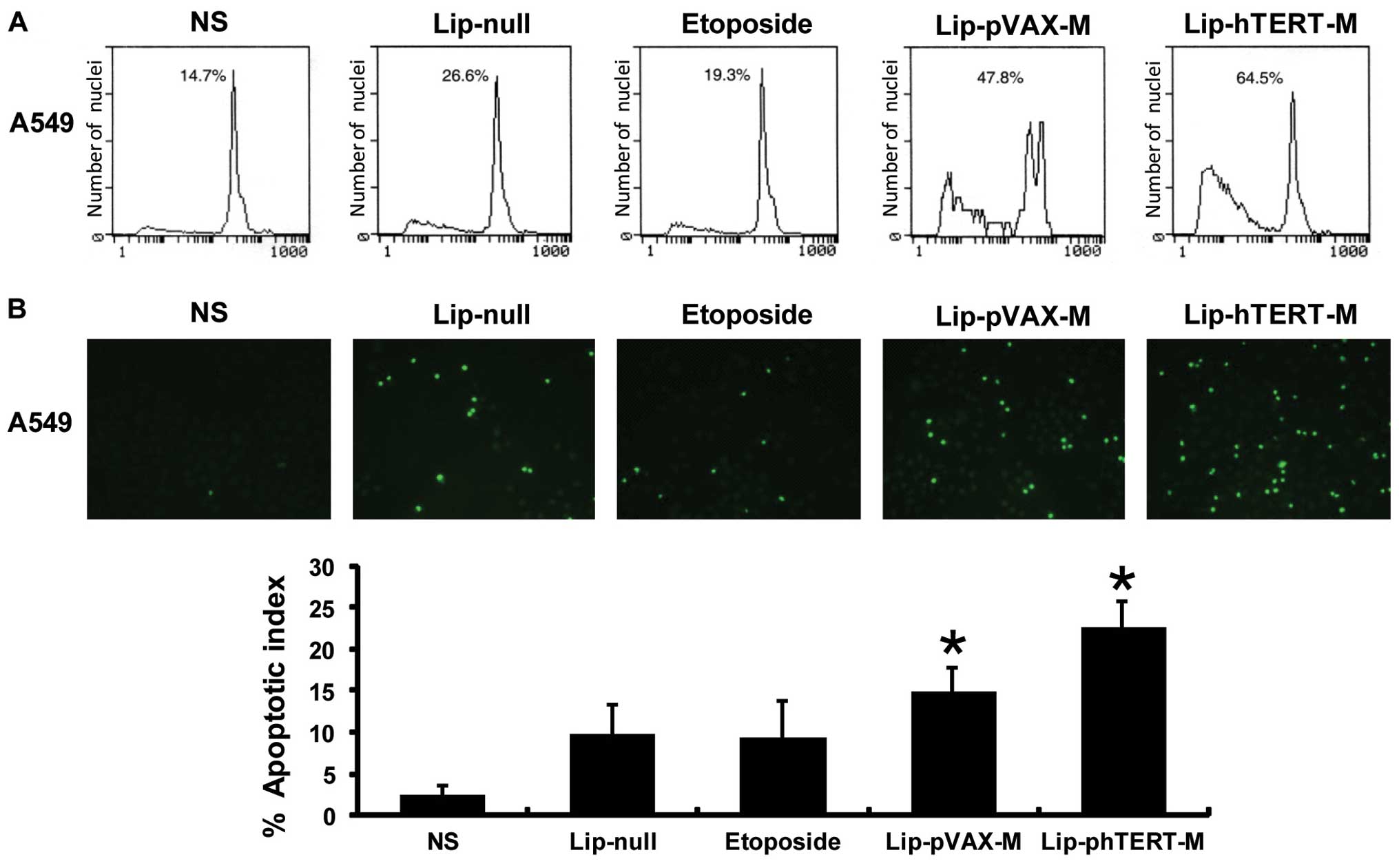
apoptotic effect of the 5 groups on A549 cells was quantitated via
flow cytometry and TUNEL assays. From the results of flow
cytometry, we revealed that the apoptotic cells accounted for 64.5%
of the cells in the Lip-phTERT-M group versus 47.8% in the
Lip-pVAX-M group, 26.6% in the Lip-null group and 14.7% in the NS
group. Furthermore, the results of the TUNEL assays suggested that
the AI had been increased the most by Lip-phTERT-M compared with
the other groups. Data are represented as the mean AI ± SD of
cancer cells, as percentage normalized to the AI of the cancer
cells (Fig. 2).
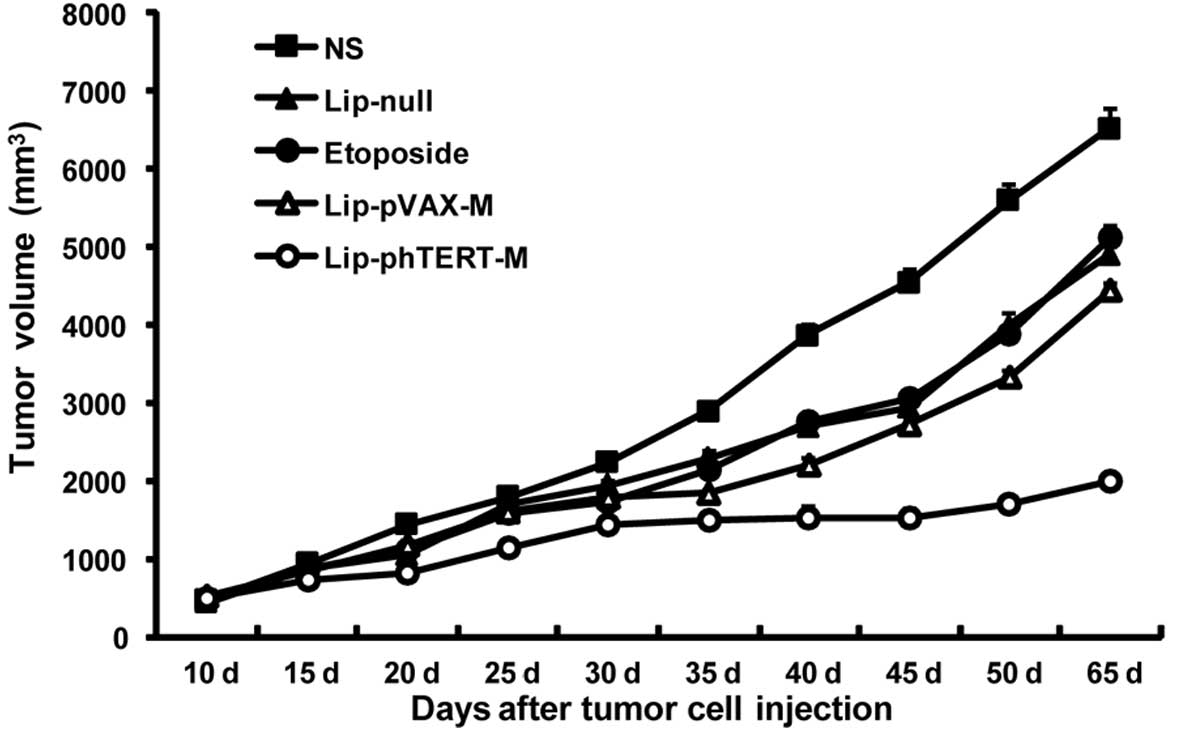
Lip-phTERT-M significantly supresses
tumor growth in the A549 tumor model
The mouse tumor model assay showed that Lip-phTERT-M
was more effective in the suppression of tumor growth than the
other groups (P<0.05). Additionally, Lip-phTERT-M resulted in
>67% inhibition of tumor growth compared with the NS group
(P<0.05, 2 days after the completion of treatment). No
significant difference in tumor volume was observed among the other
groups (Fig. 3).
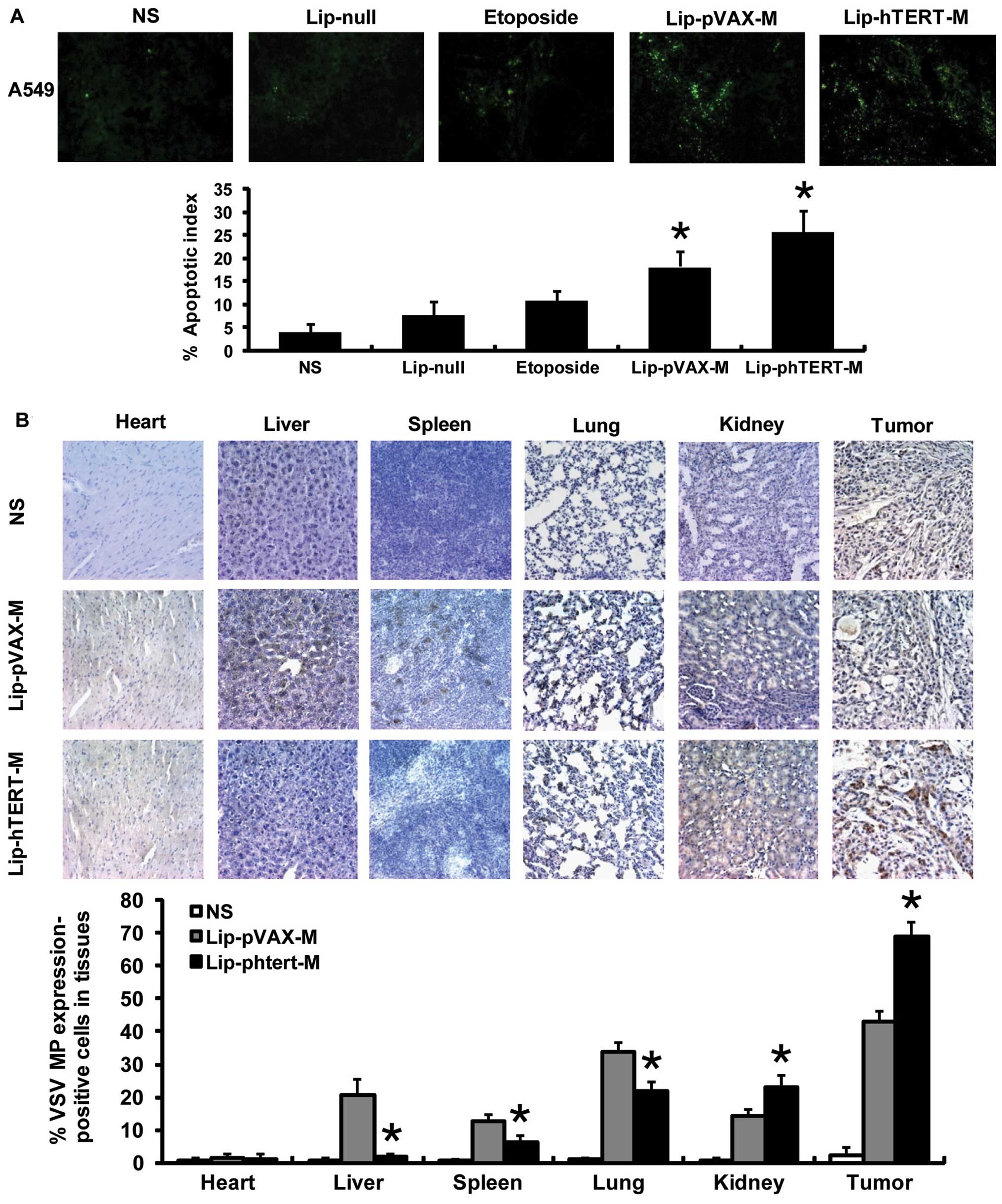
Lip-phTERT-M increases intratumoral
apoptosis in the A549 tumor model
The presence of apoptotic cells within the tumor
sections was determined by TUNEL assays. The findings demonstrated
that Lip-pVAX-M and Lip-phTERT-M enhanced the apoptotic rate of
tumor cells, and a more apparent increase in the number of
apoptotic cells was observed within the tumors of the Lip-phTERT-M
group. Data are represented as the mean AI ± SD of cancer cells, as
a percentage normalized to the AI of the cancer cells (Fig. 4A).
Lip-phTERT-M exhibits a targeted
antitumor effect on the A549 tumor model
To investigate the targeted antitumor effect of
Lip-phTERT-M, immunohistochemical staining was carried out. The
results demonstrated that Lip-phTERT-M resulted in an apparent
increase in VSV MP expression in the tumor and the kidney cells,
whereas expression in the liver, the spleen and the lung was
significantly reduced in comparison with Lip-pVAX-M (P<0.05).
Moreover, VSV MP expression in the heart in the two groups was low
and there was no significant difference between them. These
findings demonstrate that Lip-phTERT-M restricted abundant VSV MP
expression to the tumor tissues and may have a superior specific
antitumor effect to Lip-pVAX-M (Fig.
4B).
Discussion
In order to enhance the efficacy and safety of
cancer gene therapy, numerous scientific teams focus on restricting
the therapeutic gene expression to tumors. If the therapeutic gene
is expressed in all cells, it will affect tumor and normal cells. A
tumor-specific promoter system is likely to be useful for solving
this problem. However, true tumor-specific promoters are rare and
often useful only in particular types of cancer (31,32).
hTERT is the catalytic subunit of telomerase, which is highly
active in immortalized cells and >90% of human cancers but is
inactive in most normal somatic cells. It is apparently a strong
and tumor-selective promoter with potential applications in
targeted cancer gene therapy (31–33).
VSV MP induces the apoptosis of tumor cells in the
absence of other viral components without severe side-effects,
unlike VSV (21–24). Our previous studies demonstrated
that the plasmid pVAX-M alone or combined with radiation or DDP
efficiently inhibited the growth of solid tumors and significantly
prolonged survival times (25–29).
Thus, VSV MP gene therapy is a promising approach for the treatment
of tumors.
In the present study, the plasmid phTERT-M was
constructed to investigate whether it was able to inhibit tumor
growth selectively and specifically. We discovered that
Lip-phTERT-M suppressed A549 cells or the tumor model growth more
effectively by inducing a higher rate of apoptosis in A549 cells
than Lip-pVAX-M in vitro and in vivo. These findings
suggest that Lip-phTERT-M had an enhanced anti-tumor efficacy
against A549 human lung adenocarcinoma cells compared with
Lip-pVAX-M. Secondly, Lip-phTERT-M resulted in an apparent increase
in VSV MP expression in the tumor and kidney, whereas its
expression in the liver, the spleen and the lung were significantly
reduced in comparison with Lip-pVAX-M (P<0.05). These findings
may further demonstrate that Lip-phTERT-M limited the
overexpression of VSV MP to the tumor tissues and had a stronger
targeted antitumor effect on A549 models in comparison with
Lip-pVAX-M. Additionally, Lip-pVAX-M causes more abundant VSV MP
expression in the tumors than in the organs, thereby improving its
antitumor efficacy and safety. Thus, it may be proposed that the
enhanced antitumor efficacy of Lip-phTERT-M is strongly correlated
with its targeted antitumor effect.
Our data demonstrated that phTERT-M gene therapy had
an apparent targeted antitumor effect against A549 human lung
carcinoma models. Given the strong antitumor effect and minimal
toxicity, the results of our study may be of significance to the
further exploration of the potential applications of this approach
in the treatment of human lung adenocarcinoma.
Acknowledgements
This study was supported by the
National Natural Science Foundation of China (30973452), the
National Key Basic Research Program of China (973 project
2010CB529900) and the Hi-tech Research and Development Program of
China (863 project 2007AA021106).
References
|
1
|
Poole JC, Andrews LG and Tollefsbol TO:
Activity, function, and gene regulation of the catalytic subunit of
telomerase (hTERT). Gene. 269:1–12. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nakamura TM, Morin GB, Chapman KB, et al:
Telomerase catalytic subunit homologs from fission yeast and human.
Science. 277:955–959. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Greenberg RA, O’Hagan RC, Deng H, Xiao Q,
Hann SR, Adams RR, Lichtsteiner S, Chin L, Morin GB and DePinho RA:
Telomerase reverse transcriptase gene is a direct target of c-Myc
but is not functionally equivalent in cellular transformation.
Oncogene. 18:1219–1226. 1999. View Article : Google Scholar
|
|
4
|
Kanaya T, Kyo S, Hamada K, Takakura M,
Kitagawa Y, Harada H and Inoue M: Adenoviral expression of p53
represses telomerase activity through down-regulation of human
telomerase reverse transcriptase transcription. Clin Cancer Res.
6:1239–1247. 2000.
|
|
5
|
Abdul-Ghani R, Ohana P, Matouk I, Ayesh S,
Ayesh B, Laster M, Bibi O, Giladi H, Molnar-Kimber K, Sughayer MA,
et al: Use of transcriptional regulatory sequences of telomerase
(hTER and hTERT) for selective killing of cancer cells. Mol Ther.
2:539–544. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Koga S, Hirohata S, Kondo Y, Komata T,
Takakura M, Inoue M, Kyo S and Kondo S: A novel telomerase-specific
gene therapy: gene transfer of caspase-8 utilizing the human
telomerase catalytic subunit gene promoter. Hum Gene Ther.
11:1397–1406. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Koga S, Hirohata S, Kondo Y, Komata T,
Takakura M, Inoue M, Kyo S and Kondo S: FADD gene therapy using the
human telomerase catalytic subunit (hTERT) gene promoter to
restrict induction of apoptosis to tumors in vitro and in vivo.
Anticancer Res. 21:1937–1943. 2001.PubMed/NCBI
|
|
8
|
Komata T, Koga S, Hirohata S, Takakura M,
Germano IM, Inoue M, Kyo S, Kondo S and Kondo Y: A novel treatment
of human malignant gliomas in vitro and in vivo: FADD gene transfer
under the control of the human telomerase reverse transcriptase
gene promoter. Int J Oncol. 19:1015–1020. 2001.PubMed/NCBI
|
|
9
|
Komata T, Kondo Y, Kanzawa T, Hirohata S,
Koga S, Sumiyoshi H, Srinivasula SM, Barna BP, Germano IM, Takakura
M, et al: Treatment of malignant glioma cells with the transfer of
constitutively active caspase-6 using the human telomerase
catalytic subunit (human telomerase reverse transcriptase) gene
promoter. Cancer Res. 61:5796–5802. 2001.
|
|
10
|
Komata T, Kondo Y, Kanzawa T, Ito H,
Hirohata S, Koga S, Sumiyoshi H, Takakura M, Inoue M, Barna BP, et
al: Caspase-8 gene therapy using the human telomerase reverse
transcriptase promoter for malignant glioma cells. Hum Gene Ther.
13:1015–1025. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gu J, Kagawa S, Takakura M, Kyo S, Inoue
M, Roth JA and Fang B: Tumor-specific transgene expression from the
human telomerase reverse transcriptase promoter enables targeting
of the therapeutic effects of the Bax gene to cancers. Cancer Res.
60:5359–5364. 2000.
|
|
12
|
Majumdar AS, Hughes DE, Lichtsteiner SP,
Wang Z, Lebkowski JS and Vasserot AP: The telomerase reverse
transcriptase promoter drives efficacious tumor suicide gene
therapy while preventing hepatotoxicity encountered with
constitutive promoters. Gene Ther. 8:568–578. 2001. View Article : Google Scholar
|
|
13
|
Gu J, Andreeff M, Roth JA and Fang B:
hTERT promoter induces tumor-specific Bax gene expression and cell
killing in syngenic mouse tumor model and prevents systemic
toxicity. Gene Ther. 9:30–37. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li Q, Wei YQ, Wen YJ, et al: Induction of
apoptosis and tumor regression by vesicular stomatitis virus in the
presence of gemcitabine in lung cancer. Int J Cancer. 112:143–149.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gaddy DF and Lyles DS: Oncolytic vesicular
stomatitis virus induces apoptosis via signaling through PKR, Fas,
and Daxx. J Virol. 81:2792–2804. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Balachandran S and Barber GN: Vesicular
stomatitis virus (VSV) therapy of tumors. IUBMB Life. 50:135–138.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Balachandran S, Porosnicu M and Barber GN:
Oncolytic activity of vesicular stomatitis virus is effective
against tumors exhibiting aberrant p53, Ras, or myc function and
involves the induction of apoptosis. J Virol. 75:3474–3479. 2001.
View Article : Google Scholar
|
|
18
|
Stojdl DF, Lichty BD, tenOever BR, et al:
VSV strains with defects in their ability to shutdown innate
immunity are potent systemic anti-cancer agents. Cancer Cell.
4:263–275. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shinozaki K, Ebert O, Kournioti C, Tai YS
and Woo SL: Oncolysis of multifocal hepatocellular carcinoma in the
rat liver by hepatic artery infusion of vesicular stomatitis virus.
Mol Ther. 9:368–376. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ahmed M, Cramer SD and Lyles DS:
Sensitivity of prostate tumors to wild type and M protein mutant
vesicular stomatitis viruses. Virology. 330:34–49. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kopecky SA and Lyles DS: The cell-rounding
activity of the vesicular stomatitis virus matrix protein is due to
the induction of cell death. J Virol. 77:5524–5528. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kopecky SA and Lyles DS: Contrasting
effects of matrix protein on apoptosis in HeLa and BHK cells
infected with vesicular stomatitis virus are due to inhibition of
host gene expression. J Virol. 77:4658–4669. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gaddy DF and Lyles DS: Vesicular
stomatitis viruses expressing wild-type or mutant M proteins
activate apoptosis through distinct pathways. J Virol.
79:4170–4179. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ahmed M, McKenzie MO, Puckett S, Hojnacki
M, Poliquin L and Lyles DS: Ability of the matrix protein of
vesicular stomatitis virus to suppress beta interferon gene
expression is genetically correlated with the inhibition of host
RNA and protein synthesis. J Virol. 77:4646–4657. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhong Q, Wen YJ, Yang HS, et al: Efficient
inhibition of cisplatin-resistant human ovarian cancer growth and
prolonged survival by gene transferred vesicular stomatitis virus
matrix protein in nude mice. Ann Oncol. 19:1584–1591. 2008.
View Article : Google Scholar
|
|
26
|
Zhao JM, Wen YJ, Li Q, et al: A promising
cancer gene therapy agent based on the matrix protein of vesicular
stomatitis virus. FASEB J. 22:4272–4280. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Du XB, Lang JY, Xu JR, et al: Vesicular
stomatitis virus matrix protein gene enhances the antitumor effects
of radiation via induction of apoptosis. Apoptosis. 13:1205–1214.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shi W, Tang Q, Chen X, et al: Antitumor
and antimetastatic activities of vesicular stomatitis virus matrix
protein in a murine model of breast cancer. J Mol Med. 87:493–506.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Luo S, Chen P, Luo ZC, Zhang P, et al:
Combination of vesicular stomatitis virus matrix protein gene
therapy with low-dose cisplatin improves therapeutic efficacy
against murine melonoma. Cancer Sci. 101:1219–1225. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Horikawa I, Cable PL, Afshari C and
Barrett JC: Cloning and characterization of the promoter region of
human telomerase reverse transcriptase gene. Cancer Res.
59:826–830. 1999.PubMed/NCBI
|
|
31
|
Painter RG, Lanson NA Jr, Jin Z, et al:
Conditional expression of a suicide gene by the telomere reverse
transcriptase promoter for potential post-therapeutic deletion of
tumorigenesis. Cancer Sci. 96:607–613. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shieh GS, Shiau AL, Yo YT, et al: Low-dose
etoposide enhances telomerase-dependent adenovirus-mediated
cytosine deaminase gene therapy through augmentation of adenoviral
infection and transgene expression in a syngeneic bladder tumor
model. Cancer Res. 66:9957–9966. 2006. View Article : Google Scholar
|
|
33
|
Wirth T, Zender L, Schulte B, Mundt B, et
al: A telomerase-dependent conditionally replicating adenovirus for
selective treatment of cancer. Cancer Res. 63:3181–3188.
2003.PubMed/NCBI
|