Introduction
Diabetic nephropathy (DN) is the leading cause of
chronic and end-stage renal disease. It is responsible for
significant morbidity and mortality in patients with diabetes. The
production of advanced glycation end-products (AGEs) is accelerated
in diabetes and AGEs mediate the progressive alteration of renal
architecture and loss of renal function in DN (1).
The clinical signature of DN is proteinuria, which
is a marker of disease severity and an independent risk factor for
cardiovascular disease. Podocytes are the most differentiated cell
type within the glomerulus and are crucial for the maintenance of
the glomerular filtration barrier. Experimental and clinical
studies have shown that podocyte numbers are decreased in DN, and
there is a direct correlation between decreased podocyte number and
proteinuria (2). Moreover, many
podocytes in urine are viable, suggesting a primary problem was
their ability to remain attached to the underlying glomerular
basement membrane (GBM) (3). A
reduction in podocyte number may result in the failure of
mechanical support for the glomerular capillary loop, leading
directly to glomeruloscerosis (4).
Integrin-linked kinase (ILK) is important for the
control of podocyte-matrix adhesion and shape modulation. Treatment
of podocytes with the ILK inhibitor MC-5 has been shown to reduce
stress fiber formation and increase apoptosis (5). However, little is known with regard
to the effects of AGEs on ILK expression and podocyte adhesion.
It is increasingly being suggested that sustained
intrarenal renin-angiotensin system (RAS) activation is crucial in
the pathogenesis of podocyte injury and causes proteinuria. The
blockade of the RAS with angiotensin-converting enzyme inhibitors
and angiotensin II type 1 receptor (AT1R) antagonists has been
shown to reduce proteinuria and retard DN progression (6). These observations have indicated that
RAS blockade may directly affect various renal cells. Previous
studies have demonstrated that blocking the RAS restored nephrin
levels and attenuated foot process broadening in DN (7,8). The
inhibition of angiotensin II (Ang II) may directly benefit
podocytes. Our previous study (9)
showed that AGEs activated the RAS in podocytes. Accordingly, the
current study investigated whether the activated RAS was involved
in the reduction of podocyte adhesion induced by AGEs.
Materials and methods
Cell culture
A conditionally immortalized mouse podocyte cell
line was provided by Dr Peter Mundel (Harvard Medical School,
Charlestown, MA, USA). Cells were harvested as described in our
previous study (9) and cultured in
nonpermissive conditions in 10% fetal bovine serum (FBS) with 10
U/ml interferon (INF)-γ (Sigma-Aldrich, St. Louis, MO, USA) in
RPMI-1640 medium at 33°C. When the podocytes were 60–80% confluent,
the temperature was changed to 37°C and incubation was continued
for 7–10 days to allow differentiation. Passages 8–14 were used for
all experiments; prior to treatment, differentiated podocytes were
cultured in media containing 1% FBS for 24 h. Bovine serum albumin
(BSA) was purchased from MP Biomedicals (Santa Ana, CA, USA), while
AGEs were obtained from Merck (Darmstadt, Germany) and losartan was
from Sigma-Aldrich.
Experimental design
Growth-arrested podocytes were exposed to different
concentrations of AGEs (20, 40, 80 and 160 μg/ml) for 24 h prior to
cell adhesion, ILK mRNA and protein expression being measured.
Growth-arrested podocytes were pretreated for 1 h with or without
losartan (100 μM) and AGEs were subsequently added.
RNA extraction, cDNA synthesis and
quantitative polymerase chain reaction (qPCR)
Total RNA was extracted from the podocytes using
TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA, USA),
according to the manufacturer’s instructions. Total RNA (500 ng)
was reverse transcribed using reverse transcriptase (RT) in a SYBR
Premix Ex Taq kit (Perfect Real-Time; Takara Bio, Inc., Shiga,
Japan). Reactions were carried out at 37°C for 15 min and then 85°C
for 5 sec. qPCR was performed using an ABI Prism 7000 sequence
detection system (Applied Biosystems, Foster City, CA, USA) and a
SYBR Premix Ex Taq Perfect Real-Time kit (Takara Bio, Inc.). The
primers used were as follows: ILK sense, GAC GCT CAG CAG ACA TGT
GGA and anti-sense, GGA AAT ACC TGG TGG GAC GGT AG;
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) sense, AAA TGG TGA
AGG TCG GTG TGA AC and anti-sense, CAA CAA TCT CCA CTT TGC CAC
TG.
Western blot analysis
Podocytes were washed twice with cold
phosphate-buffered saline (PBS) and scraped with lysis buffer
containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 nM
Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium
pyrophosphate, 1 mM β-glycerophosphate, 1 mM
Na3VO4, 1 μg/ml leupeptin (Cell Signaling
Technology, Inc., Danvers, MA, USA) and protease and phosphatase
inhibitor cocktail tablets (Roche Diagnostics, Mannheim, Germany).
Protein concentrations were determined using the Bradford reaction.
From boiled extracts, 20 μg was loaded on 8% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels and
transferred to polyvinylidene fluoride membranes (Bio-Rad
Laboratories, Hercules, CA, USA). Membranes were blocked in 5%
fat-free milk prior to incubation with rabbit anti-ILK (1:500;
Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). Blots were
incubated with horseradish peroxidase-conjugated secondary
antibodies (Santa Cruz Biotechnology, Inc.); bands were detected
using an enhanced chemiluminescence (ECL) system (Millipore,
Billerica, MA, USA).
Cell adhesion assay
Podocytes were harvested, washed, and resuspended in
medium with different concentrations of AGEs. Equal numbers of
cells (1×105) were replated in duplicate wells of
96-well plates coated with rat-tail type I collagen (Sigma, St.
Louis, MO, USA) and allowed to attach at 37°C in a CO2
incubator for 6 h. Nonattached cells were removed by washing with
PBS twice. Attached cells were measured using a hexosaminidase
assay, as described in a previous study (5). Briefly, 3.75 mM
p-nitrophenol-N-acetyl-D-glucosaminide (Sigma) in 50 mM
citrate buffer (pH 5.0) containing 0.25% Triton X-100 was added to
each well for 1 h at 37°C. Enzyme deactivation was performed using
50 mM glycine and 5 mM EDTA (pH 10.4). Cell adhesion was quantified
using absorbance at 405 nm, measured with a Bioteck Spectramax
(Molecular Devices, Sunnyvale, CA, USA). All data were corrected
with values from control wells and experiments were repeated three
times.
Statistical analyses
Results are presented as the mean ± standard
deviation for different conditions. Statistical significance was
assessed using a nonparametric Kruskal-Wallis analysis of variance
(ANOVA) or a Student’s t-test. P<0.05 was considered to indicate
a statistically significant difference.
Results
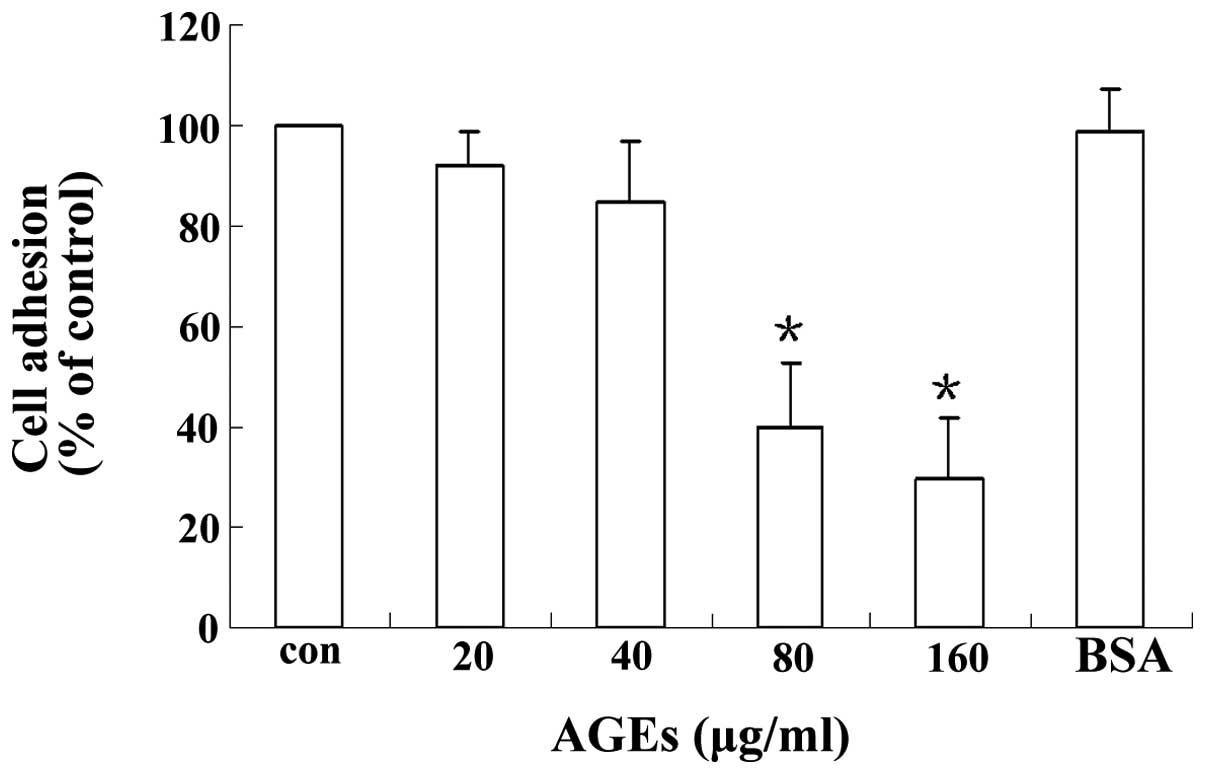
AGEs decrease podocyte adhesion
Podocytes were incubated with different
concentrations of AGEs for 24 h and podocyte adhesion was measured
using a hexosaminidase assay. As shown in Fig. 1, AGEs inhibited podocyte adhesion
in a concentration-dependent manner. Podocytes incubated with AGEs
(80 or 160 μg/ml) showed significantly inhibited adhesion compared
with cells in the control group: for 80 μg/ml, 40±13 versus 100%;
for 160 μg/ml, 30±12 versus 100% (P<0.05).
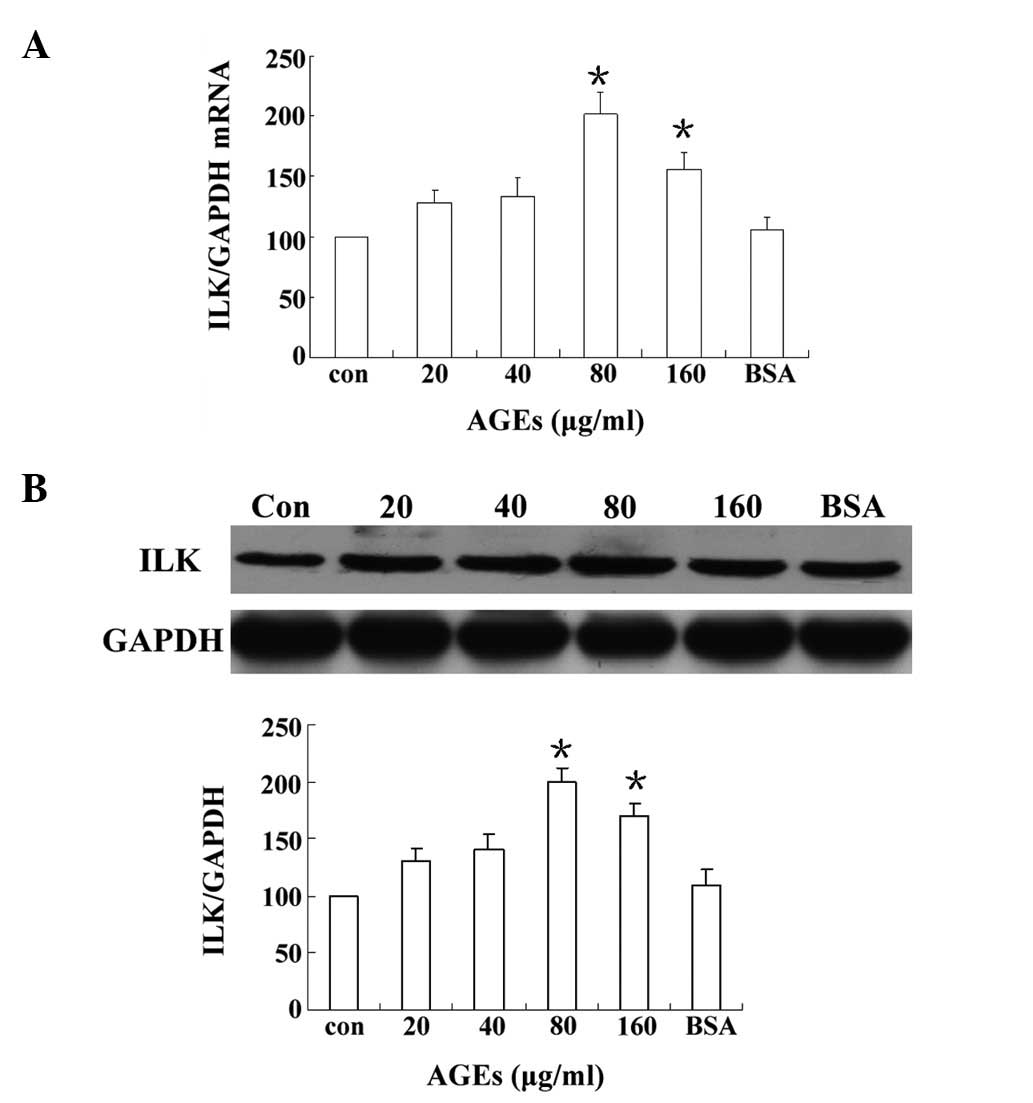
AGEs upregulate ILK mRNA and protein
levels in podocytes
To investigate the potential role of ILK in podocyte
adhesion, the effects of different concentrations of AGEs on ILK
expression in podocytes were observed. As shown in Fig. 2, ILK mRNA and protein production
were rapidly upregulated by AGEs in a concentration-dependent
manner. Concentration-response studies revealed that the maximal
expression of ILK was induced by AGEs at a concentration of 80
μg/ml. Moreover, ILK expression levels in mouse podocytes exposed
to 80 μg/ml AGEs were two-fold higher than the expression in the
control cells (Fig. 2B). A further
increase in the concentration of AGEs beyond 80 μg/ml did not
result in additional induction of ILK.
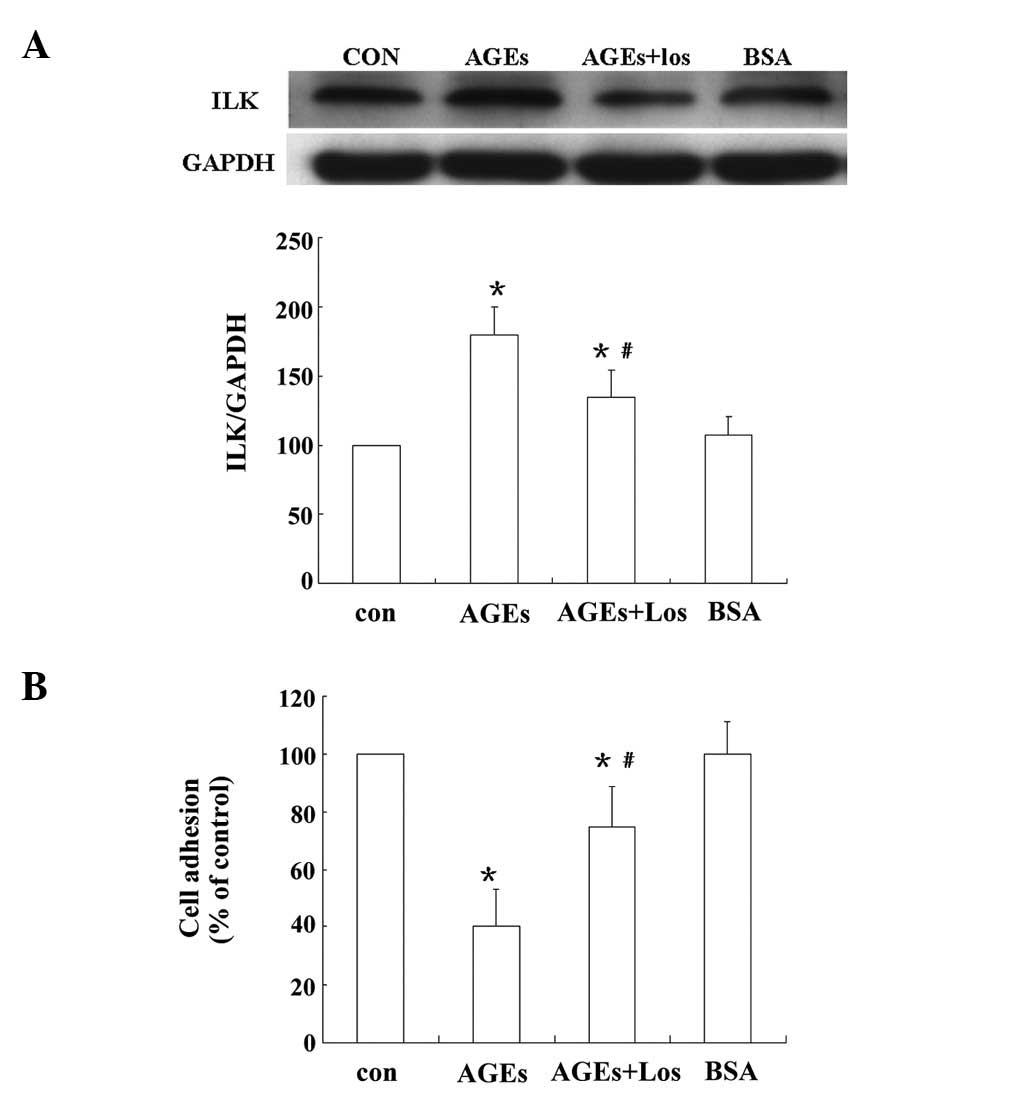
Preincubation with losartan improves the
adhesion of AGEs-treated podocytes
The effects of the angiotensin II receptor blocker
losartan on the adhesion of AGEs-treated podocytes were analyzed.
As shown in Fig. 3A, AGEs
increased ILK expression significantly; however, the AGE-induced
ILK expression was significantly inhibited by 48% following
pretreatment with losartan (P<0.05). A cell adhesion assay
revealed that, compared with control cells, adhesion was
significantly inhibited when podocytes were exposed to 80 μg/ml
AGEs (40±13 versus 100%; P<0.05). Pretreatment with losartan
(100 μM) significantly improved the podocyte adhesion compared with
that of the AGEs-treated cells (75±13 versus 40±13%; P<0.05;
Fig. 3B).
Discussion
Podocytes are a monolayer of cells on the urinary
side of the GBM; a critical number of podocytes is required for
normal functions. Focal areas in the glomerulus where the podocyte
number is reduced are vulnerable to protein loss (10). Podocytopenia is associated with the
development of glomerular sclerosis and loss of renal function,
which result from podocyte detachment, apoptosis and inability to
proliferate (10). A number of
studies (4,11) have shown that the majority of
urinary podocytes are viable and are able to attach to
collagen-coated plates. This suggests that the primary podocyte
problem in diabetes is the inhibition of cell-matrix interactions
rather than cell death. Moreover, loss of cell contact with the GBM
may trigger apoptotic death. In DN, urinary podocyte loss is a more
specific marker of ongoing glomerular damage than proteinuria
(12). Furthermore, podocyte
depletion is a major mechanism driving glomeruosclerosis. When
podocyte depletion reaches a threshold (~30%), it activates
continuous autonomous podocyte depletion until global depletion
occurs (13).
ILK has been implicated in the regulation of
numerous aspects of cellular signaling, including integrin
activation, fibronectin matrix assembly, survival and
differentiation. ILK interacts with the cytoplasmic domains of β1
and β3 integrins and mediates integrin signaling, which
participates in podocyte adhesion. However, the effects of AGEs on
ILK expression have not yet been elucidated. The results of the
current study indicated that AGEs decreased the adhesive capacity
of podocytes in a concentration-dependent manner. In addition, ILK
expression was upregulated significantly in response to AGEs.
Upregulation of ILK expression is a common response of podocytes to
injury and represents a convergent pathway that mediates podocyte
dysfunction and proteinuria. A previous study (14) demonstrated that the upregulation of
ILK expression in diabetic rats was positively correlated with
podocyte injury and albuminuria. ILK is essential in podocyte
biology. Aberrant regulation of ILK has been shown to induce
shrinkage of the podocyte cell body and elongation of the podocyte
processes (15) and is implicated
in the pathogenesis of epithelial-mesenchymal transition, an
explanation for the detachment of podocytes from the GBM (16).
The activation of the RAS is important in
progressive DN. Ang II, the final effector of the RAS, is crucial
in the pathogenesis of DN, particularly in podocyte injury. Our
previous study (9) revealed that
Ang II levels in conditioned media and cell lysates increased
significantly in podocytes exposed to AGEs compared with the levels
in a control group. To investigate whether the local RAS was
involved in the adhesion of podocytes treated with AGEs in the
present study, podocytes were pretreated with the AT1R antagonist
losartan. The results of the current study showed that pretreatment
with losartan significantly improved podocyte adhesion and
decreased ILK levels incrementally and significantly. These results
indicated that the RAS activation induced by AGEs was involved in
the inhibition of podocyte adhesion. A previous study (14) revealed that the inhibition of ILK
ameliorated the dysfunction of podocyte adhesion. Other studies
demonstrated that blocking the RAS reduced the progression of
proteinuria and nephropathy and that this effect was not explained
solely by an antihypertensive effect. The protective effect of Ang
II receptor blockade has been shown to be entirely accounted for by
a reduction in podocyte loss (13,17).
These observations suggest that RAS blockade directly affects
podocytes. Podocytes have been a focus of study, due to the fact
that they are a filtration barrier to proteins and are important in
the pathogenesis of glomerulosclerosis (18). Liebau et al(19) first demonstrated the functional
expression of key RAS components in differentiated human podocytes
using western blot analysis and immunostaining (19). Ang II is emerging as a critical
mediator of podocyte injury in diabetic kidney disease and has been
shown to cause the rearrangement of cortical F-actin,
redistribution of ZO-1, reduced α-actinin-4, dephosphorylation of
nephrin, expression of focal adhesion kinase and a migratory
phenotype switch in cultured mouse podocytes (20,21).
The results of the present study indicate that AGEs may reduce
podocyte adhesion through the upregulation of ILK expression, and
that local RAS activation in podocytes may be important in the
process.
The results of the present study indicated that the
AGEs-induced RAS activation in podocytes is important in the
pathophysiology of podocyte depletion, particularly in podocyte
adhesion. A previous study showed that collagen modified by AGEs
inhibited podocyte adhesion (22),
while another study demonstrated that AGEs inhibit podocyte
adhesion via the suppression of neuropilin 1 (NRP1) expression
(23). The findings of the present
study indicate that AGEs directly inhibit the adhesive capacity of
podocytes via the upregulation of ILK expression and the activation
of the local RAS. However, it was not possible to restore the
podocyte adhesive capacity, even when the podocytes were pretreated
with losartan, which may be due to the fact that AGEs have other
effects on podocytes, similar to those of transforming growth
factor-β1 and heparanase, which decrease the podocyte adhesive
capacity (24,25). These effects may be responsible for
podocyte damage and continuous loss in DN. The present study had
certain limitations, such as only using a conditionally
immortalized mouse podocyte cell line and a single inhibitor of
ILK, losartan. Whether these results may be extended to primarily
cultured podocytes and in vivo conditions, such as animal
models of DN induced by streptozotocin, remains to be determined.
However, the current data indicate that AGEs decreased podocyte
adhesion via the upregulation of ILK, and that angiotensin receptor
blockers may be an attractive therapeutic strategy for preventing
the pathogenic effect of AGEs.
In conclusion, the results of this study indicated
that AGEs inhibited podocyte adhesive capacity through RAS
activation and the upregulation of ILK synthesis. These results
provide important information for the largely unknown mechanism of
AGEs-mediated podocyte damage in DN. The elucidation of further
details in this signaling pathway is likely be useful for future DN
treatment choices.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81070581) and the Science
and Technology Planning Project of Guangdong Province, China (grant
nos. 2010B031600202 and 2011B080701005).
References
|
1
|
Raj DS, Choudhury D, Welbourne TC and Levi
M: Advanced glycation end products: a Nephrologist’s perspective.
Am J Kidney Dis. 35:365–380. 2000.PubMed/NCBI
|
|
2
|
Ichikawa I, Ma J, Motojima M and Matsusaka
T: Podocyte damage damages podocytes: autonomous vicious cycle that
drives local spread of glomerular sclerosis. Curr Opin Nephrol
Hypertens. 14:205–210. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nakamura T, Ushiyama C, Suzuki S, et al:
Urinary excretion of podocytes in patients with diabetic
nephropathy. Nephrol Dial Transplant. 15:1379–1383. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fogo AB: Mechanisms of progression of
chronic kidney disease. Pediatr Nephrol. 22:2011–2022. 2007.
View Article : Google Scholar
|
|
5
|
Kang YS, Li Y, Dai C, et al: Inhibition of
integrin-linked kinase blocks podocyte epithelial-mesenchymal
transition and ameliorates proteinuria. Kidney Int. 78:363–373.
2010. View Article : Google Scholar
|
|
6
|
Gross ML, El-Shakmak A, Szábó A, et al:
ACE-inhibitors but not endothelin receptor blockers prevent
podocyte loss in early diabetic nephropathy. Diabetologia.
46:856–868. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Langham RG, Kelly DJ, Cox AJ, et al:
Proteinuria and the expression of the podocyte slit diaphragm
protein, nephrin, in diabetic nephropathy: effects of angiotensin
converting enzyme inhibition. Diabetologia. 45:1572–1576. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Misfud SA, Allen TJ, Bertram JF, et al:
Podocyte foot process broadening in experimental diabetic
nephropathy: amelioration with renin-angiotensin blockade.
Diabetologia. 44:878–882. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cheng CL, Tang Y, Zheng Z, et al: Advanced
glycation end-products activate the renin-angiotensin system
through the RAGE/PI3-K signaling pathway in podocytes. Clin Invest
Med. 35:E2822012.PubMed/NCBI
|
|
10
|
Jefferson JA, Shankland SJ and Pichler RH:
Proteinuria in diabetic kidney disease: A mechanistic view point.
Kidney Int. 74:22–36. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Petermann AT, Pippin J, Krofft R, et al:
Viable podocytes detach in experimental diabetic nephropathy:
potential mechanism underlying glomerulosclerosis. Nephron Exp
Nephrol. 98:e114–e123. 2004. View Article : Google Scholar
|
|
12
|
Yu D, Petermann A, Kunter U, et al:
Urinary podocyte loss is a more specific marker of ongoing
glomerular damage than proteinuria. J Am Soc Nephrol. 16:1733–1741.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fukuda A, Wickman LT, Venkatareddy MP, et
al: Angiotensin II-dependent persistent podocyte loss from
destabilized glomeruli causes progression of end stage kidney
disease. Kidney Int. 81:40–55. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dai HY, Zheng M, Tang RN, et al:
Inhibition of integrin-linked kinase by angiotensin II receptor
antagonist, irbesartan attenuates podocyte injury in diabetic rats.
Chin Med J (Engl). 125:888–893. 2012.PubMed/NCBI
|
|
15
|
Yang Y, Guo L, Blattner SM, et al:
Formation and phosphorylation of the PINCH-1-integrin linked
kinase-alpha-parvin complex are important for regulation of renal
glomerular podocyte adhesion, architecture, and survival. J Am Soc
Nephrol. 16:1966–1976. 2005. View Article : Google Scholar
|
|
16
|
Yamaguchi Y, Iwano M, Suzuki D, et al:
Epithelial-mesenchymal transition as a potential explanation for
potential explanation for podocyte depletion in diabetic
nephropathy. Am J Kidney Dis. 54:653–664. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jennings DL, Kalus JS, Coleman CL, et al:
Combination therapy with an ACE inhibitor and an angiotensin
receptor blocker for diabetic nephropathy: a meta-analysis. Diabet
Med. 24:486–493. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Barisoni L and Mundel P: Podocyte biology
and the emerging understanding of podocyte diseases. Am J Nephrol.
23:353–360. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liebau MC, Lang D, Böhm J, et al:
Functional expression of the renin-angiotensin system in human
podocytes. Am J Physiol Renal Physiol. 290:F710–F719. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yadav A, Vallabu S, Arora S, et al: ANG II
promotes autophagy in podocytes. Am J Physiol Cell Physiol.
299:C488–C496. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hsu HH, Hoffmann S, Endlich N, et al:
Mechanisms of angiotensin II signaling on cytoskeleton of
podocytes. J Mol Med (Berl). 86:1379–1394. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pedchenko VK, Chetyrkin SV, Chuang P, et
al: Mechanism of perturbation of integrin-mediated cell-matrix
interactions by reactive carbonyl compounds and its implication for
pathogenesis of diabetic nephropathy. Diabetes. 54:2952–2960. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bondeva T, Wojciech S and Wolf G: Advanced
glycation end products inhibit adhesion ability of differentiated
podocytes in a neuropilin-1-dependent manner. Am J Physiol Renal
Physiol. 301:F852–F870. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dessapt C, Baradez MO, Hayward A, et al:
Mechanical forces and TGFbeta1 reduce podocyte adhesion through
alpha3beta1 integrin downregulation. Nephrol Dial Transplant.
24:2645–2655. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kramer A, van den Hoven M, Rops A, et al:
Induction of glomerular heparanase expression in rats with
adriamycin nephropathy is regulated by reactive oxygen species and
the renin-angiotensin system. J Am Soc Nephrol. 17:2513–2520. 2006.
View Article : Google Scholar
|