Introduction
Gelastic seizures (GSs) are known as ‘laughing
seizures’ as they may look like bouts of brief, unprovoked,
uncontrollable laughter or giggling with a facial contraction. GS
symptoms often start at an early age, occasionally even starting in
infancy. GSs often have a high frequency, occurring several times
daily. Gradually, GSs may grow worse with age, accompanied by
ensuing of other seizure types and inevitably leading to cognitive
and behavioral impediment. Although they are rarely reported, GS
cases arising from temporal or frontal regions are commonly
considered to be the typical manifestation of hypothalamic
hamartoma (HH) (1). Most cases of
gelastic seizures associated with the evolution of hypothalamic
hamartoma reported resistant to drug therapy (2), thus an early excision of the
hamartoma is required. Agenesis of the corpus callosum (ACC), an
uncommon congenital cerebral malformation, often manifests as
seizures and psychomotor retardation (3). The deficit may be complete or partial
and is difficult to detect prior to magnetic resonance imaging
(MRI) and it is usually observed in conjunction with other brain
anomalies (3).
To the best of our knowledge, only HHs associated
with complete ACC have been reported (4,5). No
studies regarding partial ACC associated with HH have been reported
with surgical outcomes, which may have different symptoms and
underlying mechanisms compared with HHs associated with complete
ACC. In this case report, we examined a rare case with a
combination of HH and partial ACC, which manifested as GSs followed
by complex partial and general seizures. We also reviewed the
literature and discussed the mechanism underlining the symptoms and
surgical outcomes.
Case report
Patient history
A 6-year-old right-handed male, had exhibited
intermittent laughter and convulsions for >5 years. The patient
was the first child of healthy parents with a non-consanguineous
marriage, and had been born uneventfully by a normal vaginal
delivery without any perinatal problems. There was no family
history of epilepsy. The seizures started when the patient was 8
months old, and manifested as brief intermittent staring followed
by uncontrollable laughter and giggling during the day and night.
The seizures lasted for a few seconds and occurred 4–10 times per
day and were initially controlled with lamotrigine (100 mg/day) and
sodium valproate (750 mg/day).
Examination
The first scalp electroencephalogram (EEG)did not
reveal any abnormal findings. Following this, the frequency of
seizures increased to 7–8 times per hour and gradually deteriorated
to GSs followed by complex partial seizures and tonic-clonic
seizures. When the patient was five years old he was admitted to
the West China Hospital of Sichuan University (Chengdu, China).
Neurological examination was conducted without observing any
pathological features, and motor and cognitive development were
found to be normal. Topiramate (100 mg/day), lamotrigine (100
mg/day) and sodium valproate (750 mg/day) were administered orally
but failed to control the deterioration of the seizures. Video-EEG
revealed that the epileptic spikes originated from the left
hemisphere, mainly the frontal and temporal regions, followed by
widespread bilateral epileptiform discharges in which the type of
spike activity differed greatly between the right and left
hemispheres. A contrast-enhanced cerebral MRI examination was
performed and revealed a well-demarcated intrahypothalamic mass
(Fig. 1A–F), which was
ovoid-shaped and located in the suprasellar region. The mass was
iso-intense to the grey matter and non-enhanced on the
post-gadolinium images (Fig. 1F).
Furthermore, the T1-weighted spin-echo (SE) (TR-450/TE-25) sagittal
slice revealed signs of partial ACC (Fig. 1D). Since the progressive epilepsy
and other symptoms may be ameliorated by surgical intervention,
early surgical excision of the HH was suggested. Following surgical
resectioning and lamina terminalis fenestration, the mass in the
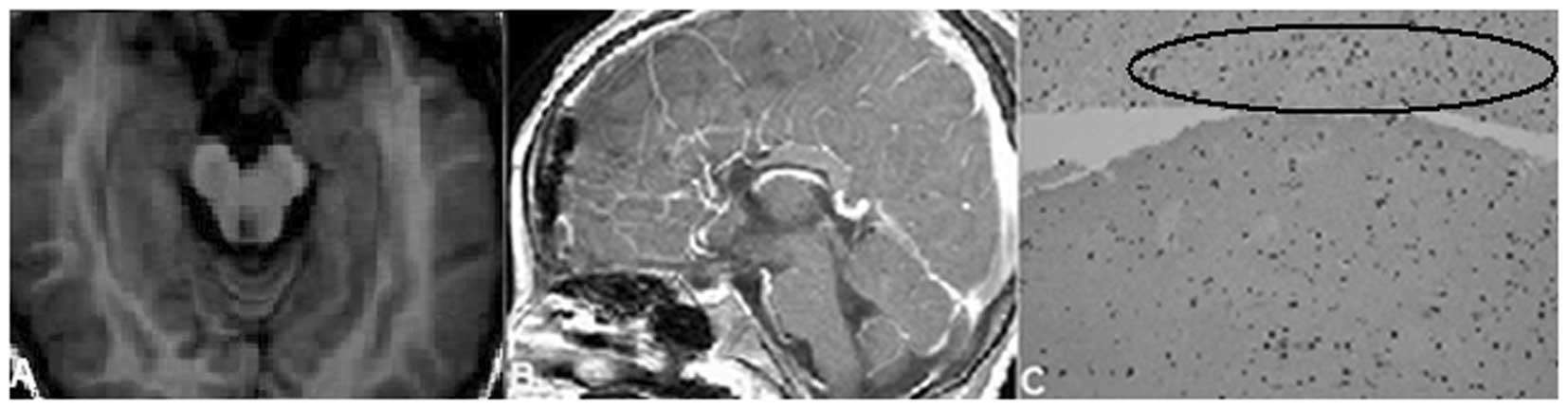
suprasellar region was completely removed (Fig. 2A and B). The mass was confirmed as
HH by post-surgical histological examination (Fig. 2C).
Follow-up
The patient was followed up at the outpatient
department of West China hospital following surgery. Anti-seizure
therapy with lamotrigine, 50mg/day, was provided following surgery.
At the one year follow-up, the seizures had been reduced to 2–4
times/day.
Discussion
HH is associated with a variety of neurological
and/or endocrinal abnormalities. The usual symptoms of a patient
with HH include GSs, precocious puberty and developmental
retardation (6). HH may be
classified into sessile (intrahypothalamic) and pedunculated
(parahypothalamic). Sessile HH is often associated with epilepsy,
while the pedunculated variety presents with precocious puberty
(7). MRI reveals the HH, which
often appears isointense to grey matter without contrast
enhancement (Fig. 1).
The nodules, or clusters of small neurons, were
considered to be the universal histological feature of HH lesions
associated with pharmacologically refractory epilepsy (2). Even rare findings such as an
independent epileptogenic focus outside of the hypothalamus were
reported. The intrinsic epileptogenicity of the HH, particularly
when the sessile HH protrudes into the third ventricle has been
previously confirmed (8).
Generally, AEDs (Antiepileptic drugs) are
ineffective for GSs associated with HH and rarely prevent cognitive
and behavioral deterioration. In a long-term follow-up retrospect
review (6 years on average), only 3 of 8 HH patients with GSs and
other types of seizures achieved acceptable control by AEDs
(9). Several studies have
demonstrated that the resection of HH is effective for long-lasting
control of seizures and may alleviate the behavioral and cognitive
abnormalities (10,11). Thus, early surgical excision of the
HH is recommended.
ACC is a cerebral malformation mainly caused by the
abnormalities of chromosomes 8, 11, 13–15 and 18 from the cells of
a fetus (12) and is often
associated with other brain anomalies such as interhemispheric
cysts with hydrocephalus, Dandy-Walker syndrome and neuronal
migrational disorder (3). The
corpus callosum functions as the anatomic connection for
transferring information between hemispheres. The defect may be
complete or partial, depending on the stage of callosal development
inhibition (3). Although ACC is
not lethal, patients with ACC may present with neurological
problems, such as complex partial seizures, intellectual impairment
and psychosis (3).
Several studies have postulated that the
deterioration of seizures with EEG abnormalities may be a form of
secondary epileptogenesis or a ‘kindling effect’ in the neocortex
(13). Although oxcarbazepine,
carbamazepine, topiramate, valproic acid and levetiracetam were
frequently used to control GSs, previous studies on HH combined
with complete ACC used conservative treatments and the number of
general seizures decreased by 75% using anticonvulsant therapy
(4,5). However, in this case we were unable
to decrease the number of refractory seizures using anticonvulsant
therapy which may be due to the differences between partial and
complete ACC as complete ACC theoretically inhibits ictal
electrical impulses transmitted to the other side of the brain. The
removal of HH would be the treatment of choice in this case.
HH and ACC patients are capable of experiencing
seizures, which in the present case may have originated from HH,
partial ACC or both. Considering the fact that the frequency of the
seizures was reduced following surgery, the seizures may have
mostly occurred from HH. However, it was considered to be unlikely
that the seizures following surgery were due to the secondary
epileptogenesis or partial agenesis of the corpus callosum, or
both. Furthermore, whether the co-occurrence of HH and ACC in our
patient was a coincidence or a new syndrome required clarification.
Clarifying this question requires further clinical evidence,
long-term clinical follow-up and genetic studies, which are likely
to be performed if a candidate gene is eventually identified.
References
|
1
|
Cascino G, Andermann F, Berkovic S, et al:
Gelastic seizures and hypothalamic hamartomas: evaluation of
patients undergoing chronic intracranial EEG monitoring and outcome
of surgical treatment. Neurology. 43:747–750. 1993. View Article : Google Scholar
|
|
2
|
Waldau B, McLendon RE, Fuchs HE, George TM
and Grant GA: Few isolated neurons in hypothalamic hamartomas may
cause gelastic seizures. Pediatr Neurosurg. 45:225–229. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Singh S and Garge S: Agenesis of the
corpus callosum. J Pediatr Neurosci. 5:83–85. 2010. View Article : Google Scholar
|
|
4
|
Chen CC, Lin YT, Chang WC, Hsieh LC and
Liang JS: Rare combination of gelastic epilepsy, agenesis of the
corpus callosum, and hamartoma. Pediatr Neurol. 45:265–267. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Alikchanov AA, Petrukhin AS, Mukhin KYu
and Nikanorov AYu: Gelastic epilepsy, hypothalamic hamartoma,
precocious puberty, and agenesis of the corpus callosum: a new
association. Brain Dev. 20:239–241. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Striano S, Santulli L, Ianniciello M,
Ferretti M, Romanelli P and Striano P: The gelastic
seizures-hypothalamic hamartoma syndrome: facts, hypotheses, and
perspectives. Epilepsy Behav. 24:7–13. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Coons SW, Rekate HL, Prenger EC, et al:
The histopathology of hypothalamic hamartomas: study of 57 cases. J
Neuropathol Exp Neurol. 66:131–141. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Téllez-Zenteno JF, Serrano-Almeida C and
Moien-Afshari F: Gelastic seizures associated with hypothalamic
hamartomas. An update in the clinical presentation, diagnosis and
treatment. Neuropsychiatr Dis Treat. 4:1021–1031. 2008.PubMed/NCBI
|
|
9
|
Castaño De La Mota C, Martín Del Valle F,
Pérez Villena A, et al: Hypothalamic hamartoma in paediatric
patients: clinical characteristics, outcomes and review of the
literature. Neurologia. 27:268–276. 2012.PubMed/NCBI
|
|
10
|
Striano S, Meo R, Bilo L, et al: Gelastic
epilepsy: symptomatic and cryptogenic cases. Epilepsia. 40:294–302.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Berkovic SF, Arzimanoglou A, Kuzniecky R,
Harvey AS, Palmini A and Andermann F: Hypothalamic hamartoma and
seizures: a treatable epileptic encephalopathy. Epilepsia.
44:969–973. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jeret JS, Serur D, Wisniewski KE and Lubin
RA: Clinicopathological findings associated with agenesis of the
corpus callosum. Brain Dev. 9:255–264. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Freeman JL, Harvey AS, Rosenfeld JV,
Wrennall JA, Bailey CA and Berkovic SF: Generalized epilepsy in
hypothalamic hamartoma: evolution and postoperative resolution.
Neurology. 60:762–767. 2003. View Article : Google Scholar : PubMed/NCBI
|