Introduction
Arsenic (As) is a naturally occurring element that
is ubiquitously present in the environment. Chronic exposure to
inorganic As has been indicated to be correlated with chronic
changes in a number of organs, including the liver, kidney, skin
and bladder. The kidney, as the primary organ for the excretion of
metabolites, appears to be one of the main targets of As (1). The toxicity of As in the kidney has
been demonstrated in the human population and animals through renal
pathology and functional changes (2–4). In
addition, the mechanisms of As-induced kidney damage have been
investigated in numerous studies (5). Among the suggested mechanisms,
oxidative stress is one of the best-accepted theories (6–8). It
has been indicated that As is capable of increasing the generation
of reactive oxygen species (ROS), such as intracellular peroxide,
superoxide anion radical (O2•−), hydrogen
peroxide (H2O2) and hydroxyl free radicals
(OH•), which stimulate proinflammatory and profibrogenic
cytokines (9) and are able to
directly or indirectly damage cellular DNA and protein (7). Therefore, ROS are a significant
causal factor in As-induced renal nephrotoxicity and fibrosis.
Transforming growth factor-β1 (TGF-β1) is a potent
profibrogenic cytokine, and elevated TGF-β1 levels are causatively
involved in the activation of profibrotic signaling pathways
initiated by oxidative stress (10). The TGF-β signaling pathway is
regulated predominantly by Smads. TGF-β-activated Smad pathways are
pivotal for the induction of extracellular matrix (ECM) generation,
myofibroblast differentiation, epithelial-mesenchymal
transformation (EMT) and disease progression (11–14).
Having reviewed the mechanisms of As-induced renal nephrotoxicity
and fibrosis, we hypothesized that scavenging ROS, reducing
oxidative stress and/or inhibiting the activities of
proinflammatory and profibrogenic cytokines may partially reduce
As-induced nephrotoxicity. Grape seed extract (GSE), which is rich
in polyphenols, has been demonstrated to possess potent antioxidant
properties and is considered to be a safe and effective antioxidant
compound. It has been shown that the antioxidant activity of GSE is
greater than that of vitamins C and E and β-carotene (15). GSE may exert its antioxidant and
anti-inflammatory effects (16) by
scavenging oxygen free radicals, inhibiting lipid peroxidation and
the formation of inflammatory cytokines, altering cell membrane
receptors and intracellular signaling pathway proteins, and
modulating gene expression (17).
In a previous study, we demonstrated that GSE may inhibit
As-induced rat liver injury (18).
The initial aim of the present study was to
elucidate whether dietary supplementation with GSE was capable of
inhibiting chronic As-induced renal injury and fibrosis. A further
aim was to explore the molecular mechanisms implicated in the
action of GSE by measuring the effects of GSE treatment on
nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox)
activity and the TGF-β signaling pathway. In this study, rats
received a life-long, non-lethal dosage of inorganic sodium
arsenite (NaAsO2, As; 30 ppm in drinking water) for 12
months, with or without the co-administration of GSE and the
effects on oxidative stress and TGF-β/Smad signaling were
evaluated.
Materials and methods
Chemicals and animal treatments
The As used in the study was purchased from Sigma
(St. Louis, MO, USA). GSE, containing monomeric catechins, dimeric
and trimeric procyanidins, and larger procyanidins (Table I), was obtained from Jianfeng, Inc.
(Lot no. G050412; Tianjin, China).
 | Table IComposition of grape seed extract
(GSE). |
Table I
Composition of grape seed extract
(GSE).
| Compounds | % (wt/wt) |
|---|
| Monomers | 3.7–3.9 |
| Catechin | 1.3–1.9 |
| Epicatechin | 1.4–2.0 |
| Epicatechin
gallate | 1.0–1.7 |
| Dimers +
trimers | 28–31 |
| Procyanidin
B1 | 6.8–7.4 |
| Procyanidin
B2 | 4.9 |
| Procyanidin
B3 | 3.2–3.7 |
| Procyanidin
B4 | 2.1 |
| Total
procyanidins | >96 |
All animal procedures were performed in accordance
with the Animal Care and Use Committee of Zhengzhou University
(Zhengzhou, China). The experimental protocols were approved by the
Animal Care and Use Committee of Zhengzhou University. The study
used healthy, male Sprague-Dawley rats (6 weeks of age; average
body weight bw, 180±10 g), which were randomly divided into four
groups (Groups 1–4). The rats in each group (10 rats per group)
were treated as follows: Group 1 received only drinking water;
Group 2 received As in the drinking water at a concentration of 30
ppm; Group 3 received 100 mg/kg GSE, which was dissolved in the
drinking water, every other day by oral gavage; and Group 4
received As plus GSE, with dosages and treatments as mentioned for
Groups 2 and 3, respectively. The animals were housed in groups of
three rats per cage at 22°C with a 12-h light/dark cycle and were
given free access to food and water. The food and water intake, as
well as the body weight of the animals, were monitored throughout
the 12-month experimental period.
Blood collection and tissue
preparation
At the end of experiment, the rats were placed in
individual metabolic cages for a 24-h urine collection. To
eliminate contamination of the urine samples, the rats received
only water during the collection period. Following the urine
collection, the animals were immediately anesthetized with ether
and the blood was collected using cardiac puncture. The blood was
allowed to clot and was subsequently centrifuged and stored at
−80°C for analysis. The renal tissues were collected and used for
protein extraction or total RNA isolation, as well as for the
analysis of ROS production or Nox activity. The protein
concentrations were assessed using a protein assay kit (Bio-Rad,
Hercules, CA, USA) and bovine serum albumin was used as a
standard.
Renal function parameters
Blood urea nitrogen (BUN), urinary protein (Up)
levels, plasma creatinine (PCr) and creatinine clearance
(CCr) were measured using commercial assay kits, in
accordance with the manufacturer’s instructions (Bioassay Systems
LLC, Hayward, CA, USA).
Oxidative damage analysis
ROS production in the renal tissues was determined
using a 2′,7′-dichlorofluorescin diacetate (DCF-DA; Invitrogen Life
Technologies, Carlsbad, CA, USA) assay, where DCF-DA was converted
into highly fluorescent DCF by cellular peroxides (including
H2O2), as previously described (19). Nox activity in the cell membranes
and cytosolic fractions was measured through the detection of ROS
production using a lucigenin-derived chemiluminescence assay with
NADPH as the substrate, as previously described (20). Protein carbonyls (PCs) were
analyzed using the 2,4-dinitrophenylhydrazine (DNPH) method, as
previously described (21). Lipid
peroxidation was assessed using a thiobarbituric acid reactive
substances (TBARS) assay (22).
All the chemicals used were analytic grade and purchased from
Sigma.
Levels of proinflammatory cytokines
Serum levels of tumor necrosis factor-α (TNF-α),
interleukin-6 (IL-6) and IL-1β were quantified using Quantikine
enzyme-linked immunosorbent assay (ELISA) kits for TNF-α, rat IL-6
and rat IL-1β (R&D Systems, Minneapolis, MN, USA),
respectively, in accordance with the manufacturer’s
instructions.
Quantitative polymerase chain reaction
(qPCR)
Total RNA was extracted from the kidney tissue using
TRIzol® reagent (Gibco, Grand Island, NY, USA). cDNA was
transcribed from 2 μg RNA using a high-capacity cDNA reverse
transcription kit (Applied Biosystems, Foster City, CA, USA), in
accordance with the manufacturer’s instructions. The target genes
were amplified using Power SYBR® Green PCR Master Mix
reagent (Applied Biosystems). The amplification was performed in a
Real-Time PCR system (Applied Biosystems 7500 systems; Applied
Biosystems) and modified PCR cycles were used, as follows: Initial
denaturation at 95°C for 2 min, followed by 35 cycles at 95°C for
30 sec and 60°C for 30 sec. The housekeeping gene β-actin was used
as an internal control, and gene-specific mRNA expression was
normalized against β-actin expression. Relative quantification
using the 2−ΔΔCT method was performed by comparisons
with the control group. The primer sequences are summarized in
Table II.
| Table IIPrimer sequences for quantitative
polymerase chain reaction. |
Table II
Primer sequences for quantitative
polymerase chain reaction.
| Gene | Forward | Reverse |
|---|
| TGF-β1 |
5′-ATACGCCTGAGTGGCTGTCT-3′ |
5′-TGGGACTGATCCCATTGATT-3′ |
| α-SMA |
5′-CCGAGATCTCACCGACTACC-3′ |
5′-TCCAGAGCGACATAGCACAG-3′ |
| FN |
5′-TGCAATGATCAGGACACCAGG-3 |
5′-GTAATTCCGGTTGCTGTACAG-3′ |
| CTGF |
5′-GAGCTTTCTGGCTGCACC-3′ |
5′-TCTCCGTACATCTTCCTG-3′ |
| β-actin |
5′-CCATTGAACACGGCATTGTC-3′ |
5′-TCATAGATGGGCACACAGTG-3′ |
Western blot analysis
Aliquots of the renal tissues were homogenized in
ice-cold lysis buffer [1% NP-40; 10% glycerol; 20 mM Tris-Cl, pH
7.5; 150 mM NaCl; 1 mM EDTA; 1 mM ethylene
glycol-O,O′-bis(2-aminoethyl)-N,N,N′,N-tetraacetic acid (EGTA), 1
mM NaVO4, 10 mM NaPO4, 10 μg/ml leupeptin and
1 mM phenylmethylsulfonyl fluoride (PMSF)]. Total protein (30–50
μg) was subjected to 12% SDS-PAGE, transferred to nitrocellulose
membranes and incubated with primary antibodies against p47phox
(sc-14015), Nox2 (sc-27635), Nox4 (sc-30141), β-actin (sc-8432),
TGF-β1 (sc-146; all from Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA), phosphorylated Smad3 (pSmad3; cat. no. 9514) or
pSmad2 (cat. no. 3101; both from Cell Signaling Technology, Inc.,
Danvers, MA, USA) overnight at 4°C. The membranes were subsequently
probed with horseradish peroxidase-coupled secondary antibodies
(Santa Cruz Biotechnology, Inc.) at room temperature for 1 h, prior
to being washed again and visualized using an enhanced
chemiluminescence reaction (Amersham ECL™ Western Blotting System;
Amersham, Piscataway, NJ, USA). The protein expression was
quantified densitometrically using LabWorks 4.5 software of
American UVP Bioimaging System (Upland, CA, USA), and changes in
expression were normalized to the internal standard, β-actin.
Statistical analysis
The grouped data were evaluated using SPSS 13.0
statistical software (SPSS, Inc., Chicago, IL, USA). The methods
used to test the hypothesis included one-way analysis of variance
(ANOVA), followed by the Least Significant Difference (LSD) test. A
value of P<0.05 was considered to indicate a statistically
significant difference. The results are expressed as the mean ±
standard deviation.
Results
GSE improves As-induced
nephrotoxicity
No rats died during the 12-month experimental
period. No significant differences were observed in the food and
water consumption among the four groups of animals. However,
As-treated rats had lower body weights and greater kidney weights
and ratios of kidney weight to body weight compared with the rats
in the control group. Moreover, chronic As exposure increased BUN,
Up and PCr levels and decreased CCr,
indicating that As treatment adversely affected renal function.
These changes were significantly attenuated in the rats by
cotreatment with GSE (Table
III).
| Table IIIGSE improves As-induced renal
injury. |
Table III
GSE improves As-induced renal
injury.
| Parameter | Control group | GSE group | As group | GSE + As group |
|---|
| BW (g) | 571.4±30.1 | 582.9±39.1 | 411.3±19.2a | 589.3±29.7b |
| KW (g) | 4.19±0.69 | 4.23±1.01 | 5.01±0.87 | 4.55±0.99 |
| KW/BW (g/kg) | 7.33±1.6 | 7.26±1.2 | 12.10±1.7a | 7.72±1.5b |
| Up (mg/day/100 g
BW) | 20.1±1.6 | 18.3±2.4 | 45.3±9.1a | 29.6±7.3ba |
| PCr
(mg/dl) | 0.68±0.09 | 0.67±0.08 | 1.90±0.15a | 1.10±0.11b |
| BUN (mg/dl) | 31.1±4.6 | 30.8±4.9 | 58.9±11.2a | 40.1±9.7b |
| CCr
(ml/min) | 4.8±0.7 | 4.9±0.9 | 2.9±0.1a | 3.9±0.4b |
GSE attenuates the As-induced production
of proinflammatory cytokines
Inflammation has been demonstrated to be important
in the initiation of tubulointerstitial injury. To gain further
insight into the mechanisms underlying the action of GSE, the
expression levels of important proinflammatory cytokines known to
be involved in the fibrotic process were studied. As shown in
Table IV, the serum levels of
IL-1β, IL-6 and TNF-α were significantly elevated in the rats in
the As-treated group as compared with those in the control group
(P<0.01). These effects, however, were suppressed by the
simultaneous administration of GSE (P<0.01), suggesting that GSE
ameliorated the As-induced hepatic inflammatory response (Table IV).
| Table IVSerum proinflammatory cytokine levels
in different treatment groups. |
Table IV
Serum proinflammatory cytokine levels
in different treatment groups.
| Cytokine | Control group | GSE group | As group | GSE + As group |
|---|
| IL-1β (pg/ml) | 17.1±6.1 | 15.3±5.7 | 58.3±17.1a | 22.9±7.2b |
| IL-6 (pg/ml) | 33.5±9.6 | 34.1±6.5 | 73.2±10.1a | 38.9±8.8b |
| TNF-α (pg/ml) | 13.9±3.3 | 11.9±2.3 | 37.7±10.1a | 16.4±9.1b |
GSE alleviates As-induced oxidative
damage
To examine whether GSE modified the ROS production
caused by chronic As exposure, renal tissue ROS production was
assessed using DCF-DA. In addition, the renal levels of lipid
peroxidation were measured according to TBARS formation, while
endogenous protein oxidation was measured according to the levels
of PCs. The results revealed that chronic As exposure resulted in
high levels of ROS, TBARS and PC production, whereas GSE treatment
significantly ameliorated these changes in As-treated rats
(Table V).
| Table VLevels of oxidative damage in the rat
kidneys in different treatment groups. |
Table V
Levels of oxidative damage in the rat
kidneys in different treatment groups.
| Oxidative stress
marker | Control group | GSE group | As group | GSE + As group |
|---|
| ROS (pmol/mg
protein) | 4.5±0.99 | 4.3±1.02 | 13.7±1.00a | 5.9±1.33b |
| TBARS (nmol/mg
protein) | 0.33±0.07 | 0.29±0.08 | 0.71±0.09a | 0.46±0.07b |
| PCs (nmol/mg
protein) | 1.66±0.36 | 1.71±0.19 | 4.50±0.45a | 2.20±0.33b |
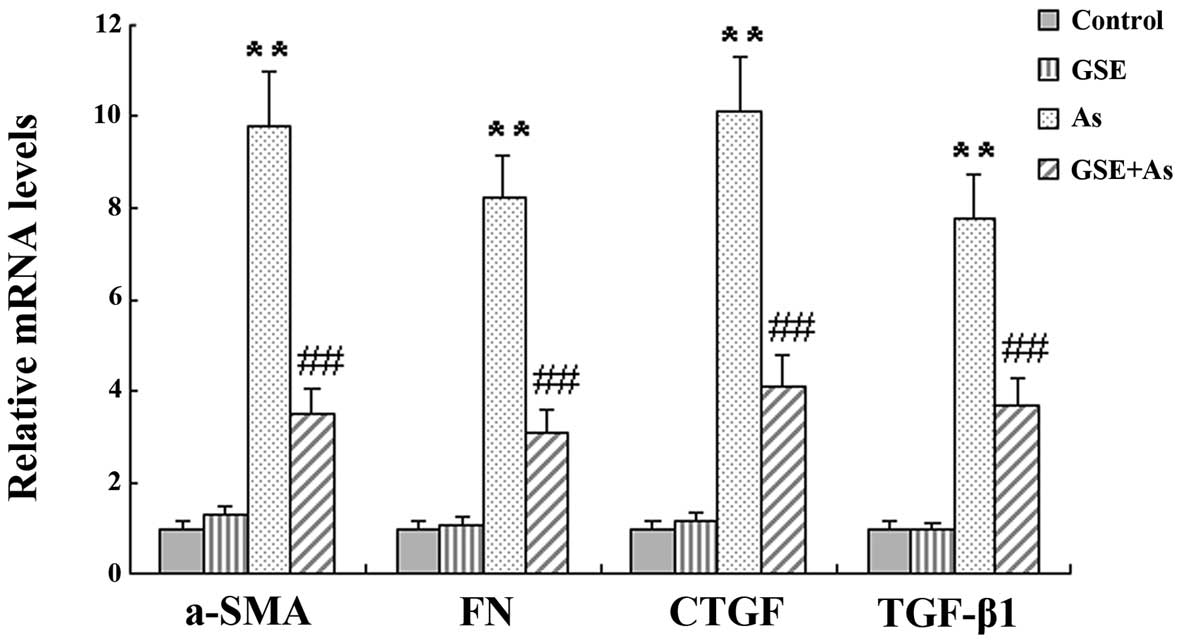
GSE attenuates As-stimulated mRNA
expression of fibrogenic genes
It has been shown that chronic As exposure induces
renal fibrosis. In the present study, this was corroborated by the
quantification of the mRNA levels of various profibrogenic genes,
specifically, α-smooth muscle actin (α-SMA), TGF-β1, connective
tissue growth factor (CTGF) and fibronectin (FN). However, GSE
cotreatment significantly attenuated the changes in the
profibrogenic gene mRNA levels in the As-treated rat liver tissues
(Fig. 1).
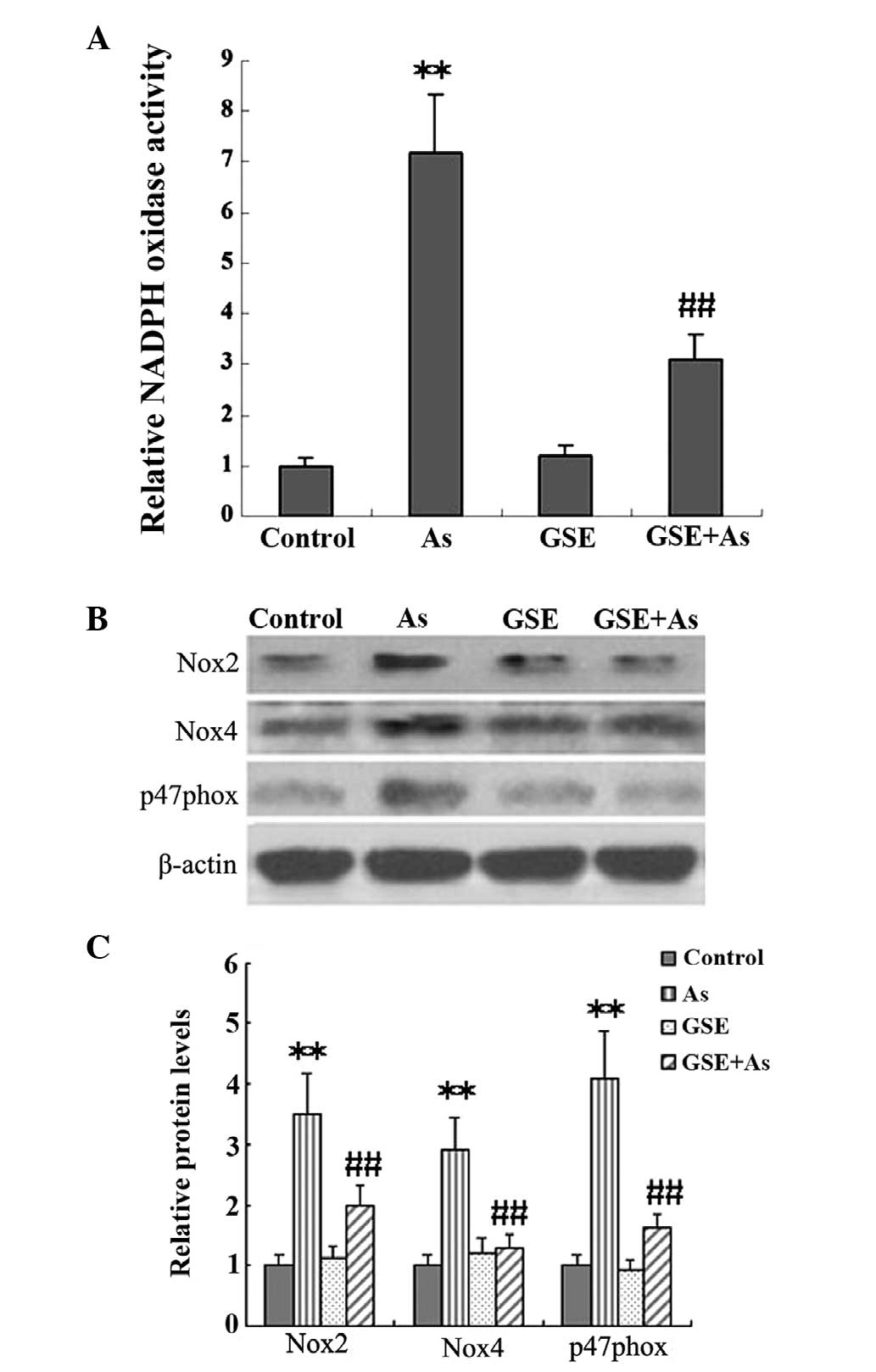
GSE inhibits As-induced Nox
It has been demonstrated that NADPH-derived ROS
generation is pivotal in the progression of renal fibrosis.
Therefore, in the present study, Nox activity and the protein
expression levels of the NADPH subunits were assessed in kidney
tissues, and the effects of GSE cotreatment on these variables were
studied. The Nox activity and protein expression levels of the
NADPH subunits, Nox2, p47phox and Nox4, were significantly elevated
in the As-treated rats as compared with the controls, and these
increases were significantly alleviated by GSE cotreatment
(Fig. 2).
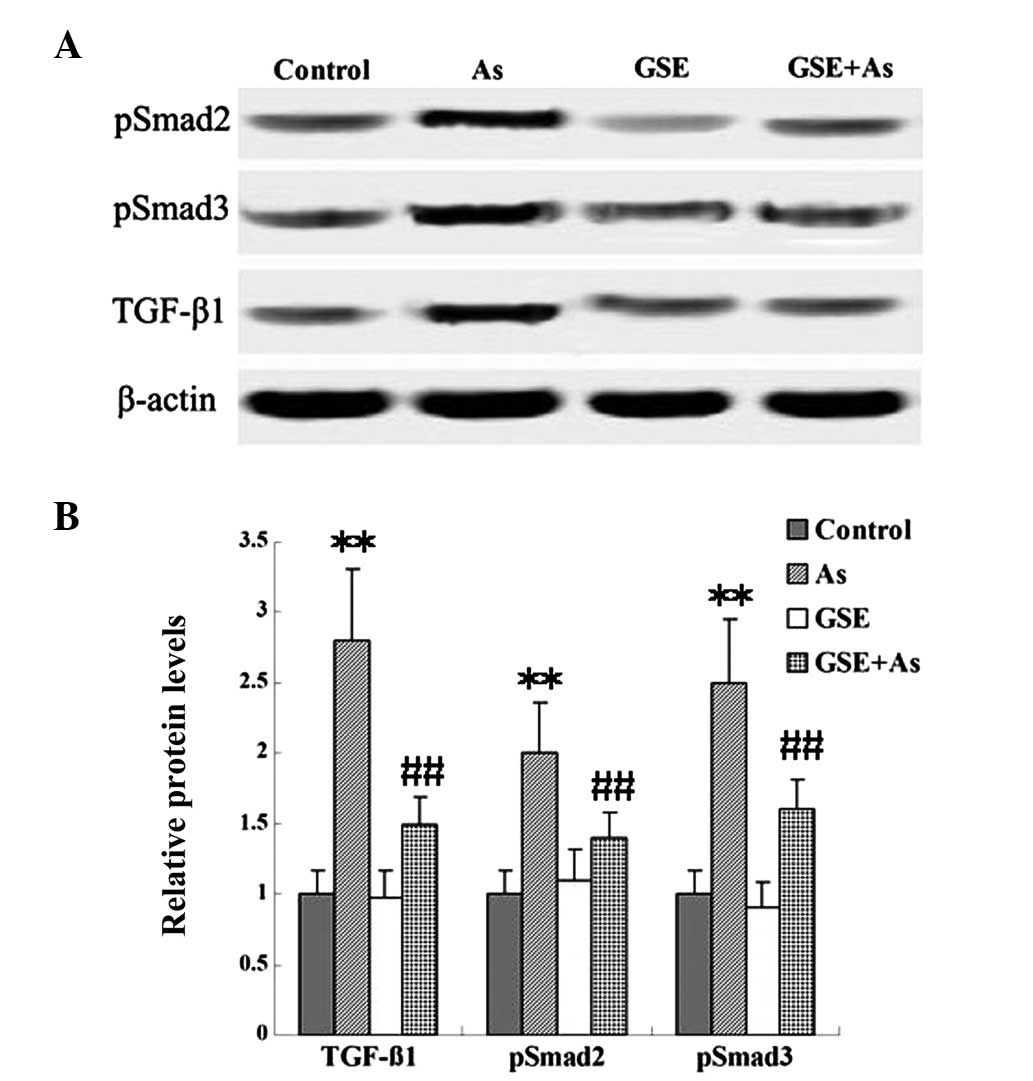
GSE decreases As-induced TGF-β1/Smad
signaling
TGF-β is an important signal transduction pathway
mediator for renal fibrogenesis, which mediates its profibrotic
effects by activating receptor-associated Smads (Smad2 and Smad3).
In the present study, the protein levels of TGF-β1 and pSmad2/3
were examined in renal tissues. As demonstrated using western
blotting, chronic As-administration caused marked increases in the
expression of TGF-β1 and pSmad2/3 when compared with the expression
levels in the control rats; however, cotreatment with GSE
attenuated these changes (Fig.
3).
Discussion
In the present study, chronic As exposure was
demonstrated to lead to renal dysfunction, as demonstrated by the
increased BUN and PCr and decreased CCr
levels (Table III). There is a
common consensus that ROS and proinflammatory and profibrogenic
cytokines, which result in oxidative damage, are important in the
progression of fibrosis (23).
Thus, it was plausible to hypothesize that the supplementation of
an antioxidant during As treatment was likely to reduce As-induced
renal fibrosis (24). GSE, which
is rich in catechin, epicatechin and procyanidin, possesses greater
antioxidant activity than vitamins C and E, as well as β-carotene
(15), and is considered to be a
safe and effective antioxidant compound. The antioxidant and
anti-inflammatory capacity of GSE has been previously demonstrated
in other tissues (25,26). The present study has, for the first
time, to the best of our knowledge, demonstrated the protective
effect of GSE against nephrotoxicity in rats following chronic As
exposure at a low concentration. It was shown that cotreatment with
GSE significantly improved renal function, attenuated As-induced
serum levels of proinflammatory cytokines and prevented kidney
fibrosis. Moreover, GSE treatment was capable of reducing
As-stimulated NADPH-derived ROS generation. In addition, it was
revealed that GSE cotreatment led to a significant reduction in
TGF-β/Smad signaling.
In the present study, a significant increase in the
level of ROS production in the As treatment group indicated that
oxidative damage was implicated in the nephrotoxicity caused by As
(Table V). Exposure to As has been
shown to lead to the increased generation of ROS (27–29).
Furthermore, out of numerous pathways, Nox has been suggested to be
the major source of ROS generation (30,31),
as demonstrated by the increased Nox activity and protein levels of
NADPH subunits, Nox2, p47phox and Nox4, in the present study.
However, cotreatment with GSE led to a pronounced recovery in the
As-induced oxidative injury (Fig.
2). These results were indicative of the antioxidant effect of
GSE.
Chronic As exposure resulted in oxidative stress in
the renal tissues. Oxidative stress may have caused further lipid
peroxidation (Table V), directly
damaging the membranes of cells and organelles and leading to the
release of reactive aldehydes with potent proinflammatory (Table IV) and profibrotic (Fig. 1) properties. This, in turn, may
have promoted oxidative stress. TGF-β1, as the most potent
profibrogenic cytokine, is responsible for matrix synthesis by
mesenchymal cells, such as fibroblasts, in vitro and during
renal fibrosis (32,33). TGF-β1 accelerates renal fibrosis by
a number of mechanisms. TGF-β1 stimulates fibroblasts to convert
into myofibroblasts, increases the expression of α-SMA and FN and
increases the synthesis of ECM components (34). Furthermore, TGF-β1 inhibits the
activity of a variety of ECM-degrading enzymes, such as matrix
metalloproteinases (MMPs) and plasminogen activator, thereby
inhibiting the degradation of the ECM. In addition, TGF-β1 may
stimulate tubular epithelial myofibroblast transdifferentiation
(35,36). The TGF-β signaling pathway is
regulated predominantly by Smads. TGF-β binds to a receptor on the
cell surface, forming a complex of subunits known as transforming
growth factor-β receptor 1 (TGFR1) and TGFR2. TGFR1 and TGFR2
activate serine/threonine kinases that subsequently mediate
signaling through the Smad family of transcriptional activators
(37,38). It has been demonstrated that
Smad2/3 is phosphorylated by activated TGFR1 and inhibited by
Smad7. Moreover, Smad7 is capable of increasing the
ubiquitin-mediated degradation of TGFR1 itself, thus preventing
TGF-β signal transduction. The results of the present study
indicated that the As-induced rat renal injury was correlated with
TGF-β1-induced fibrosis, as demonstrated by the increased levels of
TGF-β1 and pSmad2/3 in renal tissue. GSE cotreatment attenuated
these changes (Fig. 3).
Based on the results of the present study, it is
hypothesized that the enhanced production of ROS and
proinflammatory and profibrogenic cytokines may have been the
possible mechanisms underlying the As-induced oxidative stress,
which was critical in the development and progression of renal
fibrosis. In addition, the results demonstrate that the activation
of TGF-β/Smad signaling was correlated with As-induced renal
fibrosis, and that the suppression of TGF-β/Smad activation was
involved in the beneficial effects of GSE. In conclusion, GSE, a
powerful antioxidant with diverse beneficial effects, may be a
promising agent for the prevention of renal fibrosis and
dysfunction caused by chronic As exposure at a low concentration in
drinking water.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant nos. 30500540 and 81170630) and
the Program for New Century Excellent Talents in University (grant
no. NCET-09-0123).
Abbreviations:
|
NADPH
|
nicotinamide adenine dinucleotide
phosphate
|
|
BUN
|
blood urea nitrogen
|
|
CCr
|
creatinine clearance
|
|
PCr
|
plasma creatinine
|
|
α-SMA
|
α-smooth muscle actin
|
|
FN
|
fibronectin
|
|
CTGF
|
connective tissue growth factor
|
|
GSE
|
grape seed extract
|
|
ROS
|
reactive oxygen species
|
|
Nox
|
NADPH oxidase
|
|
PCs
|
protein carbonyls
|
|
TBARS
|
thiobarbituric acid reactive
substances
|
References
|
1
|
Bao L and Shi H: Potential molecular
mechanisms for combined toxicity of arsenic and alcohol. J Inorg
Biochem. 104:1229–1233. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Eom SY, Lee YC, Yim DH, Lee CH, Kim YD,
Choi BS, Park CH, Yu SD, Kim DS, Park JD and Kim H: Effects of
low-level arsenic exposure on urinary N-acetyl-β-D-glucosaminidase
activity. Hum Exp Toxicol. 30:1885–1891. 2011.
|
|
3
|
Chen JW, Chen HY, Li WF, Liou SH, Chen CJ,
Wu JH and Wang SL: The association between total urinary arsenic
concentration and renal dysfunction in a community-based population
from central Taiwan. Chemosphere. 84:17–24. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen Y, Parvez F, Liu M, et al:
Association between arsenic exposure from drinking water and
proteinuria: results from the Health Effects of Arsenic
Longitudinal Study. Int J Epidemiol. 40:828–835. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kitchin KT: Recent advances in arsenic
carcinogenesis: modes of action, animal model systems, and
methylated arsenic metabolites. Toxicol Appl Pharmacol.
172:249–261. 2001. View Article : Google Scholar
|
|
6
|
Jomova K, Jenisova Z, Feszterova M, Baros
S, Liska J, Hudecova D, Rhodes CJ and Valko M: Arsenic: toxicity,
oxidative stress and human disease. J Appl Toxicol. 31:95–107.
2011.PubMed/NCBI
|
|
7
|
Kitchin KT and Ahmad S: Oxidative stress
as a possible mode of action for arsenic carcinogenesis. Toxicol
Lett. 137:3–13. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kitchin KT and Conolly R: Arsenic-induced
carcinogenesis - oxidative stress as a possible mode of action and
future research needs for more biologically based risk assessment.
Chem Res Toxicol. 23:327–335. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brunati AM, Pagano MA, Bindoli A and
Rigobello MP: Thiol redox systems and protein kinases in hepatic
stellate cell regulatory processes. Free Radic Res. 44:363–378.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Samarakoon R, Overstreet JM, Higgins SP
and Higgins PJ: TGF-β1→SMAD/p53/USF2→PAI-1 transcriptional axis in
ureteral obstruction-induced renal fibrosis. Cell Tissue Res.
347:117–128. 2012.
|
|
11
|
Liu Y: Renal fibrosis: new insights into
the pathogenesis and therapeutics. Kidney Int. 69:213–217. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu Y: New insights into
epithelial-mesenchymal transition in kidney fibrosis. J Am Soc
Nephrol. 21:212–222. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Iwano M and Neilson EG: Mechanisms of
tubulointerstitial fibrosis. Curr Opin Nephrol Hypertens.
13:279–284. 2004. View Article : Google Scholar
|
|
15
|
Bagchi D, Bagchi M, Stohs SJ, Das DK, Ray
SD, Kuszynski CA, Joshi SS and Pruess HG: Free radicals and grape
seed proanthocyanidin extract: importance in human health and
disease prevention. Toxicology. 148:187–197. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li WG, Zhang XY, Wu YJ and Tian X:
Anti-inflammatory effect and mechanism of proanthocyanidins from
grape seeds. Acta Pharmacol Sin. 22:1117–1120. 2001.PubMed/NCBI
|
|
17
|
Kris-Etherton PM, Lefevre M, Beecher GR,
Gross MD, Keen CL and Etherton TD: Bioactive compounds in nutrition
and health-research methodologies for establishing biological
function: the antioxidant and anti-inflammatory effects of
flavonoids on atherosclerosis. Annu Rev Nutr. 24:511–538. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pan X, Dai Y, Li X, Niu N, Li W, Liu F,
Zhao Y and Yu Z: Inhibition of arsenic induced-rat liver injury by
grape seed exact through suppression of NADPH oxidase and
TGF-β/Smad activation. Toxicol Appl Pharmacol. 254:323–331.
2011.PubMed/NCBI
|
|
19
|
Morris EM, Whaley-Connell AT, Thyfault JP,
Britton SL, Koch LG, Wei Y, Ibdah JA and Sowers JR: Low aerobic
capacity and high fat diet contributes to oxidative stress and
IRS-1 degradation in the kidney. Am J Nephrol. 30:112–119. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hu R, Wang YL, Edderkaoui M, Lugea A, Apte
MV and Pandol SJ: Ethanol augments PDGF-induced NADPH oxidase
activity and proliferation in rat pancreatic stellate cells.
Pancreatology. 7:332–340. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Draper HH and Hadley M: Malondialdehyde
determination as index of lipid peroxidation. Methods Enzymol.
186:421–431. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Levine RL, Garland D, Oliver CN, Amici A,
Climent I, Lenz AG, Ahn BW, Shaltiel S and Stadtman ER:
Determination of carbonyl content in oxidatively modified proteins.
Methods Enzymol. 186:464–478. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Barnes JL and Gorin Y: Myofibroblast
differentiation during fibrosis: role of NAD(P)H oxidases. Kidney
Int. 79:944–956. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Flora SJ, Chouhan S, Kannan GM, Mittal M
and Swarnkar H: Combined administration of taurine and monoisoamyl
DMSA protects arsenic induced oxidative injury in rats. Oxid Med
Cell Longev. 1:39–45. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dulundu E, Ozel Y, Topaloglu U, Toklu H,
Ercan F, Gedik N and Sener G: Grape seed extract reduces oxidative
stress and fibrosis in experimental biliary obstruction. J
Gastroenterol Hepatol. 22:885–892. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Terra X, Montagut G, Bustos M, Llopiz N,
Ardèvol A, Bladé C, Fernández-Larrea J, Pujadas G, Salvadó J, Arola
L and Blay M: Grape-seed procyanidins prevent low-grade
inflammation by modulating cytokine expression in rats fed a
high-fat diet. J Nutr Biochem. 20:210–218. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mishra D and Flora SJ: Differential
oxidative stress and DNA damage in rat brain regions and blood
following chronic arsenic exposure. Toxicol Ind Health. 24:247–256.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bhadauria S and Flora SJ: Response of
arsenic-induced oxidative stress, DNA damage and metal imbalance to
combined administration of DMSA and monoisoamyl-DMSA during chronic
arsenic poisoning in rats. Cell Biol Toxicol. 23:91–104. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nandi D, Patra RC and Swarup D: Oxidative
stress indices and plasma biochemical parameters during oral
exposure to arsenic in rats. Food Chem Toxicol. 44:1579–1584. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Suzuki S, Arnold LL, Pennington KL,
Kakiuchi-Kiyota S and Cohen SM: Effects of co-administration of
dietary sodium arsenite and an NADPH oxidase inhibitor on the rat
bladder epithelium. Toxicology. 261:41–46. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Straub AC, Clark KA, Ross MA, Chandra AG,
Li S, Gao X, Pagano PJ, Stolz DB and Barchowsky A:
Arsenic-stimulated liver sinusoidal capillarization in mice
requires NADPH oxidase-generated superoxide. J Clin Invest.
118:3980–3989. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Okuda S, Languino LR, Ruoslahti E and
Border WA: Elevated expression of transforming growth factor-beta
and proteoglycan production in experimental glomerulonephritis:
Possible role in expansion of the mesangial extracellular matrix. J
Clin Invest. 86:453–462. 1990. View Article : Google Scholar
|
|
33
|
Gaedeke J, Peters H, Noble NA and Border
WA: Angiotensin II, TGF-beta, and renal fibrosis. Contrib Nephrol.
135:153–160. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bondi CD, Manickam N, Lee DY, Block K,
Gorin Y, Abboud HE and Barnes JL: NAD(P)H oxidase mediates
TGF-beta1-induced activation of kidney myofibroblasts. J Am Soc
Nephrol. 21:93–102. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Okada H, Danoff TM, Kalluri R and Neilson
EG: Early role of Fsp1 in epithelial-mesenchymal transformation. Am
J Physiol. 273:F563–F574. 1997.PubMed/NCBI
|
|
36
|
Fan JM, Ng YY, Hill PA, Nikolic-Paterson
DJ, Mu W, Atkins RC and Lan HY: Transforming growth factor beta
regulates tubular epithelial-myofibroblast transdifferentiation in
vitro. Kidney Int. 56:455–1467. 1999.PubMed/NCBI
|
|
37
|
Attisano L and Wrana JL: Signal
transduction by the TGF-beta superfamily. Science. 296:1646–1647.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Schnaper HW, Hayashida T and Poncelet AC:
It’s a Smad world: Regulation of TGF-beta signaling in the kidney.
J Am Soc Nephrol. 13:1126–1128. 2002.
|