Introduction
Angiocentric glioma (AG) is a rare type of tumor of
the central nervous system (1). It
was recognized as a tumor with distinct clinicopathological
characteristics by the World Health Organization (WHO) in 2007
[International Classification of Diseases for Oncology (ICD-O)
9431/1, WHO level I]. The exact cause of this disease is unclear.
The diagnosis of AG depends on tissue pathology, due to a lack of
marked specific changes with brain computed tomography (CT).
Magnetic resonance imaging (MRI) reveals a characteristic high
T2-weighted image (WI) (2) and
fluid-attenuated inversion recovery (FLAIR) signals (3,4),
while enhanced scanning reveals no enhancement (5,6).
According to clinical findings, most patients with AG suffer from
intractable epilepsy. The majority of simple surgical excisions of
the lesions result in a favorable prognosis, and postoperative
pathology has indicated that most lesions are WHO level I.
A total of 52 cases of AG have been described in the
literature (2,3,5–20),
including the current case [three cases (21) have not been included in this total,
due to a lack of specificity]. The majority of the case reports
have focused on the imaging, histopathology and clinical
manifestations of AG, while there has been less focus on studying
the intraoperative conditions and methods for the surgical excision
of the lesion. However, studies of this nature may be beneficial in
establishing how to control the symptoms of epilepsy and are worthy
of further investigation.
We report a case of seven-year-old female patient
who, according to the preoperative presentation, appeared to be
suffering from drug-refractory epilepsy. Lesions were located near
the central sulcus in the right parietal cortex, and were
concurrent with a left temporal pole arachnoid cyst. Intraoperative
cortical electroencephalogram (ECoG) monitoring was used to assist
with locating the epileptic foci, prior to the tumor and
epileptogenic lesions being surgically resected. A postoperative
study was conducted into the cytology and immunohistochemistry and
the presence of AG (WHO level I) was demonstrated pathologically.
The majority of the tumors described previously have been distinct
entities; few have been combined with cystic degeneration and a
simultaneously occurring left temporal arachnoid cyst. These are
manifestations that cause a clinical concern for the neurosurgeon.
We used intraoperative ECoG monitoring to locate the epileptic
foci, prior to surgically resecting the tumor and epileptogenic
lesions. This was beneficial for the control of postoperative
epileptic seizures. This study may be used as a reference for
clinical neurosurgeons. The case is described in the following
sections.
Case report
General information
A seven-year-old girl presented at The Department of
Neurosurgery, Yuquan Hospital, Tsinghua University (Beijing, China)
with a three-year history of paroxysmal convulsions with loss of
consciousness and a deterioration of the disease. The predominant
forms of seizure were absence and whole body tonic-clonic seizures,
while the main clinical presentation was a sudden bow (slightly
forwards and downwards, to the left) in a single or continuous
cluster of seizures. The body sometimes dipped slowly to the left
and then to the right side while the upper and lower limbs
stiffened, presenting absence seizures. The seizures were
maintained for approximately half a minute (not >1 min), prior
to remission. The administration of oral drugs, such as
oxcarbazepine, clonazepam, topiramate and valproate, controlled the
symptoms poorly and the attacks occurred 4–10 times monthly. There
was no previous history of dystocia, hypoxia, encephalitis or any
other medical history. Written informed consent was obtained from
the patient’s family.
Assisted examination
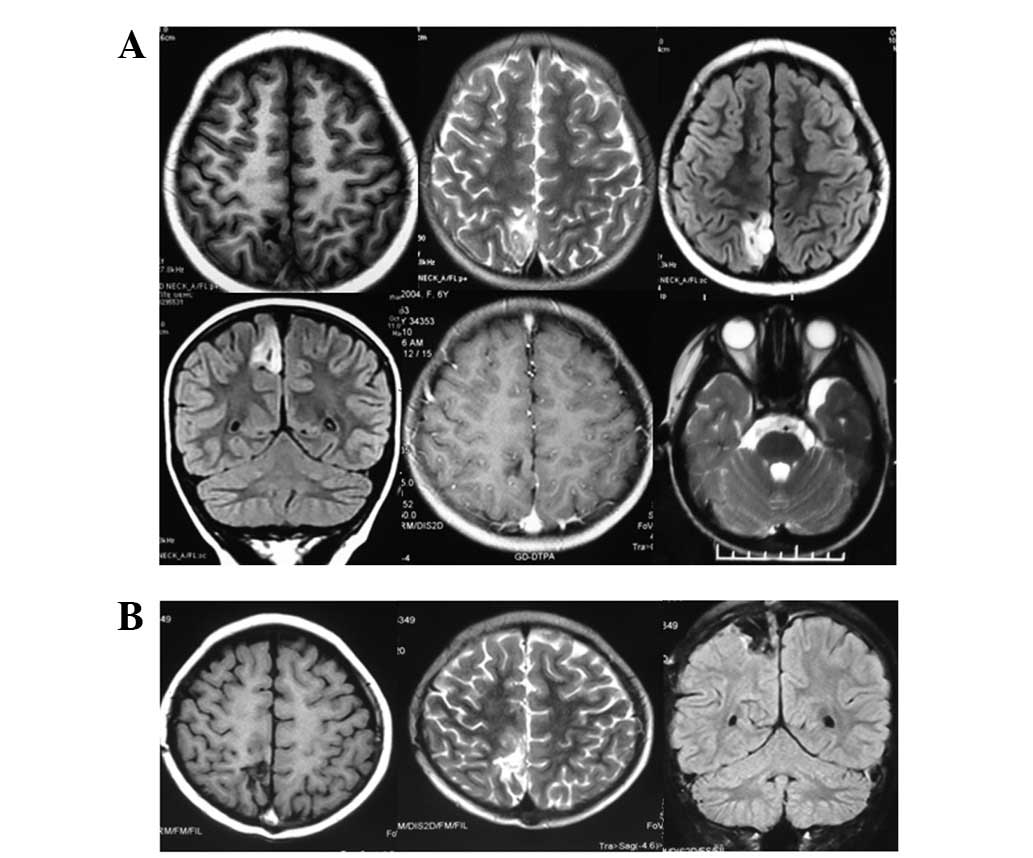
Brain MRI imaging revealed a clearly visible
boundary in the right parietal region of the cerebral falx and the
cortex. It appeared as a wedge, with the cutting-edge pointing to
the white matter of deep lesions. The T1-WI had a low signal (long
T1), while the T2-WI had a high signal (long T2) and FLAIR imaging
showed a high signal, with a low signal in the center. No
enhancement signals were detected by enhanced scanning. A visibly
widened subarachnoid cyst was observed in the left temporal pole.
Postoperatively, it was considered that there was an embryonic
dysplastic neuroepithelial tumor and left temporal pole arachnoid
cyst (Fig. 1).
The various periods of the bilateral sleep-wake
cycle led to a large number of high volatility waves. Video
electroencephalogram (VEEG) showed 1.2–2.5 Hz sharp wave and sharp
slow wave (distributing or continuously appearing), which were
obvious in bilateral central and front temporal regions. The
occurrence in right side was a little earlier than left side.
During the process of monitoring, the seizures occurred several
times and the clinical onset was predominantly a sudden bow
(slightly forwards and downwards to the left). Seizures occurred
singly or in continuous clusters. EEG concurrently displayed the
highest conductivity of the high-amplitude biphasic slow waves or
irregular slow waves on the right side.
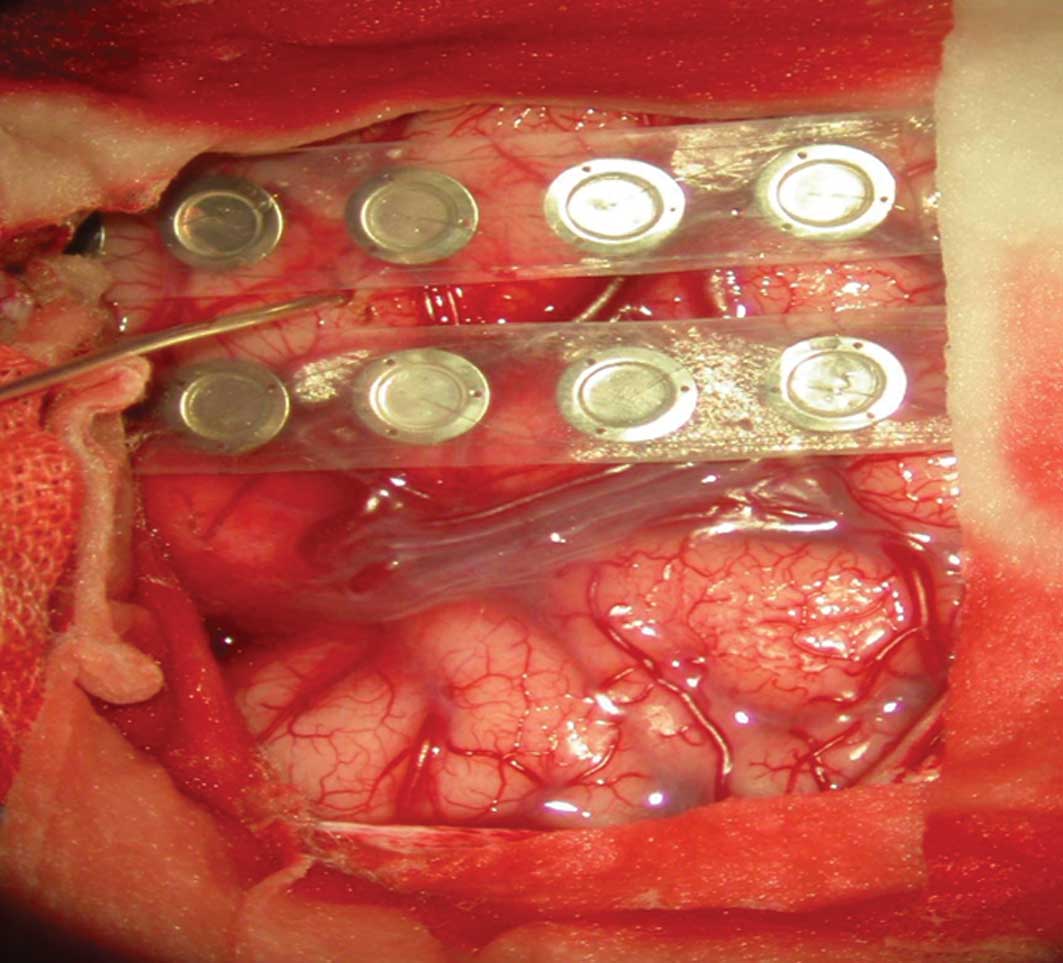
Surgery
Under general anesthesia, an incision was made in
the right parietal lesions. During the surgery, local arachnoid
thickening and subarachnoid widening were observed by cutting the
dura mater. Intraoperative ECoG and deep EEG were monitored
(Fig. 2).
The results demonstrated that local and deep spikes
were emitted frequently and that fewer were recorded 1 cm away from
the lesion edge. A few sharp wave and spike wave were visible in
areas close to posterior part of precentral gyrus and postcentral
gyrus. The lesions were located in cortical and subcortical
regions; the size was ~2.5×3.0 cm and clear borders with the
surrounding brain tissue were visible. Parts of the lesion were
textured like a rotten fish and local central monitoring with the
ECoG was repeated to reveal a small amount of spike-wave discharge
in the central frontal gyrus on the midline at ~1 cm. There were no
significantly abnormal EEGs monitored after using local cortical
low power, bipolar coagulation and burning.
Tissue pathology and the
histopathological examination
The histological examination revealed that bipolar
single cells in a monolayer or multilayers were centered around the
cortical blood vessels in the form of ependymoma-like
pseudorosettes along the vascular axis. The tumor cells were
arranged in palisade-like structures in a swirling effect, giving
the area of the tumor in the neural parenchyma a wide range of
different cell densities. The nuclei of the tumor cells were
elongated, the chromatin was finely granular, no or rare nuclear
fission was present and no microvascular proliferation or necrosis
was observed. There were no significantly heteromorphic neurons in
the lesions.
The tumor cells were positive for tumor cell glial
fibrillary acidic protein (GFAP), S-100 protein, vimentin, human
leukocyte differentiation antigen 34 (CD34), epithelial membrane
antigen (EMA), cluster of differentiation 99 (CD99) and D2-40.
However, the cells were negative for neuron markers, such as
synaptophysin (Syn) and chromaffin protein/NeuN. There was no
expression of oligodendrocyte transcriptor-2 (Olig-2), epidermal
growth factor receptor (EGFR), neurofilament (NF), CD117, nestin or
microtubule-associated protein 2 (MAP2) in the tumor cells.
Furthermore, there were no visible changes in the p53 expression
level. The Ki-67/MIBk-1 labeling index was ~1% (Fig. 3).
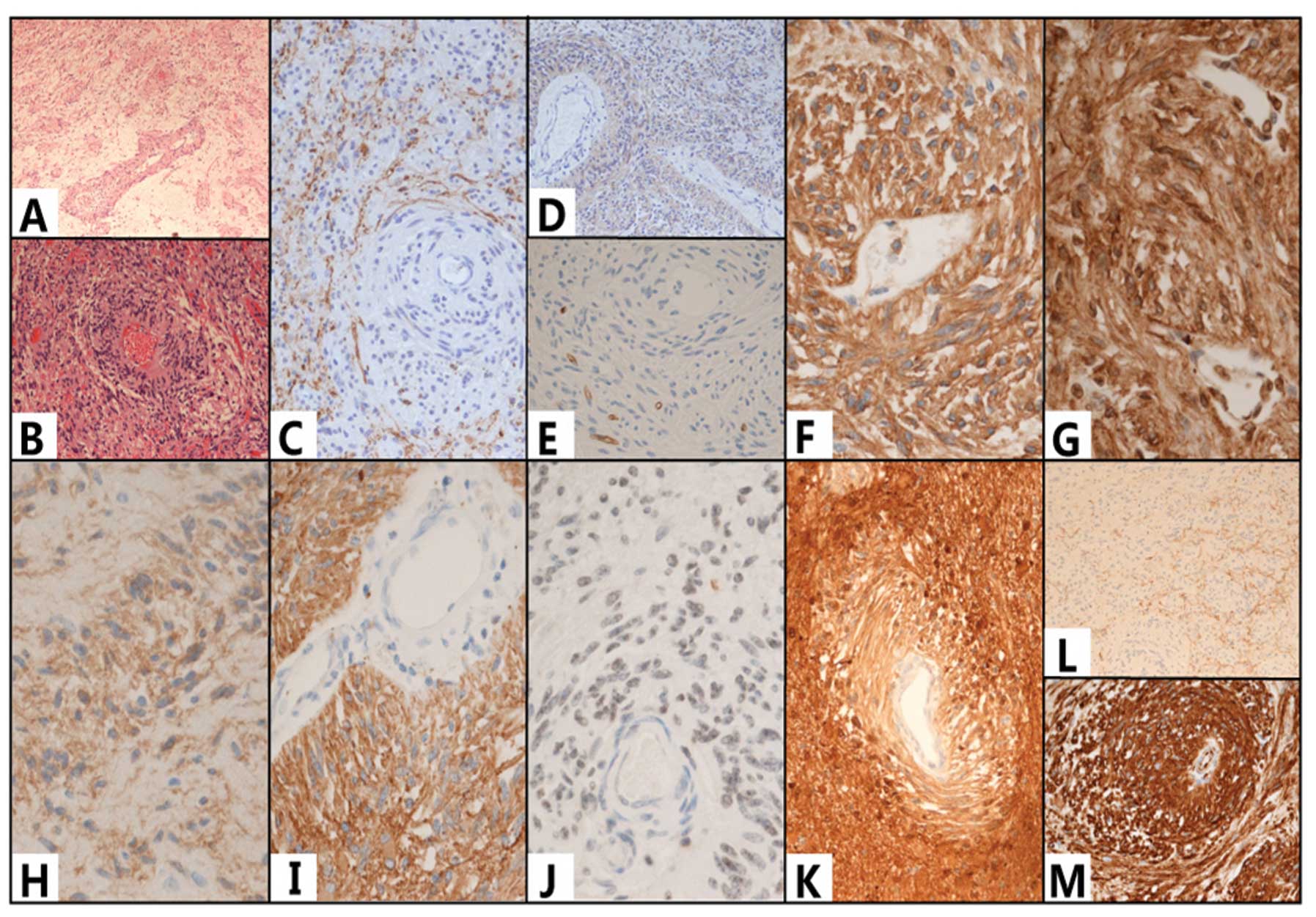 | Figure 3(A and B) Hematoxylin and eosin
(H&E) staining showed that a large number of bipolar cells were
clustered around and growing along the long axis of the the blood
vessel. The cells were positive for human leukocyte differentiation
antigen 34 (CD34) (E), CD99 (F) and D2-40 (G), glial fibrillary
acidic protein (GFAP) (I), S-100 protein (K) and vimentin (M);
however, they were negative for the neuron markers synaptophysin
(Syn) (L) and chromaffin protein/NeuN. There was no expression of
microtubule-associated protein 2 (MAP2) (C), nestin (D), epidermal
growth factor receptor (EGFR) or (H) oligodendrocyte transcriptor-2
(Olig-2). The was expression of (J) epithelial membrane antigen
(EMA) but no neurofilament (NF) in the tumor cells. (A)
Magnification, ×10; (B–D,K,L) magnification, ×20; (E–J,M)
magnification, ×40. |
Follow-up
The recovery of the patient was good following
surgery and follow-ups were performed for 12 months. No nervous
system dysfunction, such as epilepsy attacks, dizziness/headaches
or hemiplegia, was observed and there were no abnormal
somatosensory manifestations. MRI review did not reveal any
residual tumor or tumor recurrence. No abnormal EEG signals were
detected in the EEG review.
Discussion
AG is a rare type of tumor with distinct
clinicopathological characteristics. To date, there have been 52
cases of AG described in total (2,3,5–20),
including the current case [three cases (21) have not been included in this total,
due to a lack of specificity]. Morbidity due to AG may occur at any
age and seizure is the main characteristic feature of the
condition. The tumor develops slowly and may be cured by surgical
resection.
A search of the PubMed index indicates that 52 cases
of AG, including the current case, have been reported to date. The
patients were aged between two (2)
and 70 (6) years at the time of
surgery and included: children (0–6 years old), 26.92% (14/52);
juveniles (7–17 years old), 46.15% (24/52); young adults (18–46
years old), 21.15% (11/52); middle-aged adults (45–59 years old),
3.85% (2/52) and the elderly (aged >60 years), 1.92% (1/52). The
course of the disease ranged from one week (23) to 57 years (6), while the male to female ratio of AG
in the reported cases is 30:22. Therefore, it may be concluded that
the majority of patients with AG and seizures are children and
juveniles, with no significant differences in gender.
The combined analysis of the reported cases revealed
that epilepsy was a predominant clinical manifestation for the
majority of patients with AG, including 46 cases of intractable
epilepsy (90.2%, 46/51; one of the studies (12) did not describe the symptoms and so
was not included in the analysis). In 40 cases (8.43%, 40/51) the
epilepsy manifested alone; in three cases (5.88%, 3/51) this was
accompanied by symptoms of headache and dizziness; in two cases
(17) (3.92%, 2/51) by symptoms of
visual impairment and in one case (16) (1.96%, 1/51) by auditory
hallucinations. In four cases (7.84%, 4/51) headache and dizziness
manifested separately, while unsteady gait and hydrocephalus
manifested separately in one case (11) (1.96%, 1/51). It has been revealed
that epilepsy caused by a tumor accounts for ~17.8% of cases and
that 20% of the tumors are AGs (21). AGs account for 2.3% of the cases of
tumor-induced epilepsy in children (22).
MRI is beneficial when determining the location of
the lesions and in making the diagnosis. Out of all described
cases, MRI showed lesions in the frontal lobe in 11 cases (21.57%,
11/51); in the parietal lobe in seven cases (13.73%, 7/51); in the
temporal lobe in 19 cases (37.25%, 19/51); in the occipital lobe in
two cases (3.92%, 2/51) and in the hippocampus in four cases
(7.84%, 4/51). Furthermore, there was one case (1.96%, 1/51) of a
lesion in the occipitoparietal lobe, three cases (5.88%, 3/51) in
the frontoparietal lobe, one case (1.96%, 1/51) in the
frontotemporal lobe, one case (1.96%, 1/51) in the occipital
temporal lobe, one case (1.96%, 1/51) in the thalamus (17) and one case (1.96%, 1/51) in the
midbrain (11) in the junctional
zone. The imaging shows that the tumors occurred mainly in the
brain parenchyma and in each brain lobe. No coverage in the
cerebral ventricle was observed. The common imaging features are
that the brain tumor is typically located cortically or
subcortically and forms wedge-shaped lesions, which diffusely
infiltrate the surface in a stem-like (stalk-like) manner in the
direction of the cerebral ventricle (1,4). The
T2-WI and FLAIR imaging show high signals, while no enhancement
signals are shown by enhanced scanning. (5,6).
However, low (2) and high signals
(3,4) may be identified on the T1-WI. The
present case in our hospital showed a low signal T1-WI. At present,
it is unclear whether the signal changes in the T1-WI are
associated with bleeding (13) or
calcification (4,17).
Although AG has been newly recognized as a distinct
entity of tumor, compared with ependymoma (in particular, cortical
or subcortical ependymoma), there are a number of similarities in
clinical presentation and histopathology. However, AG has
characteristic features, including mild visible atypia and local
invasion of single and bipolar cells, as shown under the
microscope. Furthermore, the cells of the tumor cluster around
vessels and grow along the long axis of blood vessels in the
network of nerve fibers. Immunohistochemistry has shown the tumor
cells to be positive for GFAP (1),
EMA (23), monoclonal antibody of
cell proliferation-associated nuclear antigen (MIB-1) (10) and cell cycle-associated nuclear
antigen Ki-67. In the present case, immunohistochemistry revealed
that the tumor was positive for GFAP, S-100 protein, vimentin,
CD34, CD99 and D2-40 and negative for neuron markers (Syn and
chromaffin protein/NeuN). Furthermore, there was no expression of
Olig-2, EGFR, NF, CD117 or MAP2 in the tumor cells and there were
no visible changes in the p53 expression level. The Ki-67/MIBk-1
labeling index was ~1% (Fig. 3).
The characteristic antibodies of glial and epithelial cells were
expressed; however, the cells were negative for neuronal
antibodies. In the current case there was no change in the
expression of the proto-oncogene, p53, and the Ki-67/MIBk-1
labeling index demonstrated that the proliferation rate of the
tumor was ~1%, which is consistent with the biological behavior of
benign tumors.
AG, defined as WHO level I, has benign biological
characteristics. The main purpose of therapy is to surgically
excise the lesion and to control the seizures. Following the
surgical excision of the lesions, the symptoms of epilepsy may be
effectively controlled. Out of the 52 patients with AG, 34 patients
underwent a complete surgical resection only (received surgery but
not radiotherapy or chemotherapy) and 22 of the patients were
followed-up for >12 months without suffering any seizures; nine
patients underwent a partial resection (surgery plus radiotherapy
and chemotherapy not included) and of the seven patients who
received follow-ups for >12 months, two were seizure-free. These
data show that full surgical resection of the tumor is more
efficacious at controlling the symptoms of epilepsy. In the present
case, intraoperative ECoG monitoring showed that the abnormal
discharge range exceeded the area of the tumor, demonstrating that
resection of the tumor alone does not completely eliminate the
abnormal discharge. Since this may lead to epileptic seizures,
surgical ECoG positioning may be used to determine the area
requiring removal and to extend the target for resection, in order
to ensure normal function. In the current study, low-power
coagulation and cautery was performed, in order to obtain better
clinical efficacy for the functional areas of the cortex that were
unresected, but where abnormal discharge remained. This suggests
that for AG, surgery and intraoperative stimulation, in the central
area when necessary, are able to better ensure the efficacy of the
surgery, with a smaller impact on the patient’s function. Previous
studies have shown that in cases of AG with surrounding cortical
dysplasia (17,19,21),
intraoperative ECoG monitoring is necessary to ensure the complete
resection of all of the epileptic foci, in addition to the tumor.
In cases where the tumors have a deep location or when patients are
unwilling to accept surgery, radiotherapy alone may be applied;
however, it is necessary to initially biopsy the lesion, in order
to confirm the pathological diagnosis.
The prognosis of the patients who have undergone
total surgical resection for AG is typically good. The longest
follow-up of patients with AG has reached 165 months, and the
patients have been seizure-free in this period. Two patient
mortalities have been reported: One patient (8) succumbed to bronchitis a week
subsequent to surgery and one patient (7) succumbed to malignant tumors after 62
months. The slow growth rate of AG is generally <1%; but growth
rates have been observed to reach 10% (14). It is increasingly being
demonstrated that the cells in AG undergo active mitotic division
and have a high proliferative capacity (18,20).
Whether this may change the benign biological characteristics of
the tumor remains inconclusive. However, the long-term follow-up
for the outcome of this disease is very important.
The main clinical manifestation of AG is epilepsy.
The imaging studies of AG exhibit clear features; however, a
definite diagnosis depends on histopathological examination. The
most effective treatment approach for the control of the epileptic
seizures is to perform a total resection of the tumor and the area
presenting with abnormal discharge using intraoperative ECoG
positioning. Following the total resection to remove the lesions,
the prognosis of the majority of the patients is favorable;
however, a long-term follow-up of the patients after the surgery is
required.
References
|
1
|
Louis DN, Ohgaki H, Wiestler OD, et al:
The 2007 WHO classification of tumours of the central nervous
system. Acta Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fulton SP, Clarke DF, Wheless JW, et al:
Angiocentric glioma-induced seizures in a 2-year-old child. J Child
Neurol. 24:852–856. 2009.PubMed/NCBI
|
|
3
|
Rho GJ, Kim H, Kim HI and Ju MJ: A case of
angiocentric glioma with unusual clinical and radiological
features. J Korean Neurosurg Soc. 49:367–369. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Amemiya S, Shibahara J, Aoki S, et al:
Recently established entities of central nervous system tumors:
review of radiological findings. J Comput Assist Tomogr.
32:279–285. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lellouch-Tubiana A, Boddaert N, Bourgeois
M, et al: Angiocentric neuroepithelial tumor (ANET): a new
epilepsy-related clinicopathological entity with distinctive MRI.
Brain Pathol. 15:281–286. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Preusser M, Hoischen A, Novak K, et al:
Angiocentric glioma: report of clinico-pathologic and genetic
findings in 8 cases. Am J Surg Pathol. 31:1709–1718. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang M, Tihan T, Rojiani AM, et al:
Monomorphous angiocentric glioma: a distinctive epileptogenic
neoplasm with features of infiltrating astrocytoma and ependymoma.
J Neuropathol Exp Neurol. 64:875–881. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Arsene D, Ardeleanu C, Ogrezeanu I and
Danaila L: Angiocentric glioma: presentation of two cases with
dissimilar histology. Clin Neuropathol. 27:391–395. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lum DJ, Halliday W, Watson M, et al:
Cortical ependymoma or monomorphous angiocentric glioma?
Neuropathology. 28:81–86. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sugita Y, Ono T, Ohshima K, et al: Brain
surface spindle cell glioma in a patient with medically intractable
partial epilepsy: a variant of monomorphous angiocentric glioma?
Neuropathology. 28:516–520. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Covington DB, Rosenblum MK, Brathwaite CD
and Sandberg DI: Angiocentric glioma-like tumor of the midbrain.
Pediatr Neurosurg. 45:429–33. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sun FH, Piao YS, Wang W, et al: Brain
tumors in patients with intractable epilepsy: a clinicopathologic
study of 35 cases. Zhonghua Bing Li Xue Za Zhi. 38:153–157.
2009.(In Chinese).
|
|
13
|
Hu XW, Zhang YH, Wang JJ, et al:
Angiocentric glioma with rich blood supply. J Clin Neurosci.
17:917–918. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ma X, Ge J, Wang L, et al: A 25-year-old
woman with a mass in the hippocampus. Brain Pathol. 20:503–506.
2010.PubMed/NCBI
|
|
15
|
Mott RT, Ellis TL and Geisinger KR:
Angiocentric glioma: a case report and review of the literature.
Diagn Cytopathol. 38:452–456. 2010.
|
|
16
|
Rosenzweig I, Bodi I, Selway RP, et al:
Paroxysmal ictal phonemes in a patient with angiocentric glioma. J
Neuropsychiatry Clin Neurosci. 22:118–123. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Marburger T and Prayson R: Angiocentric
glioma: a clinicopathologic review of 5 tumors with identification
of associated cortical dysplasia. Arch Pathol Lab Med.
135:1037–1041. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pokharel S, Parker JR Jr, Parker J, et al:
Angiocentric glioma with high proliferative index: case report and
review of the literature. Ann Clin Lab Sci. 41:257–261.
2011.PubMed/NCBI
|
|
19
|
Takada S, Iwasaki M, Suzuki H, et al:
Angiocentric glioma and surrounding cortical dysplasia manifesting
as intractable frontal lobe epilepsy - case report. Neurol Med Chir
(Tokyo). 51:522–526. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li JY, Langford LA, Adesina A, et al: The
high mitotic count detected by phospho-histone H3 immunostain does
not alter the benign behavior of angiocentric glioma. Brain Tumor
Pathol. 29:68–72. 2012. View Article : Google Scholar
|
|
21
|
Prayson RA: Tumours arising in the setting
of paediatric chronic epilepsy. Pathology. 42:426–431. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Prayson RA, Fong J and Najm I: Coexistent
pathology in chronic epilepsy patients with neoplasms. Mod Pathol.
23:1097–1103. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lehman NL: Central nervous system tumors
with ependymal features: a broadened spectrum of primarily
ependymal differentiation? J Neuropathol Exp Neurol. 67:177–188.
2008. View Article : Google Scholar : PubMed/NCBI
|