Introduction
The characterization of tumor-associated antigens
(TAAs), recognized by cellular or humoral effectors of the immune
system, has provided novel perspectives for cancer therapy
(1). Immunomodulation strategies,
such as peptide-based approaches or gene vaccines, are considered
to be potential adjuvant therapies in patients with cancer, either
to treat minimal residual disease or to prevent relapse. These
strategies are based on the hypothesis that the T-cell repertoire
of an individual contains TAA-primed memory T cells, and that the
patient’s immune system is capable of being sensitized to the TAAs
of the patient’s own tumor (2).
The lysosome-associated protein transmembrane 4β
(LAPTM4B) gene contains two translation initiation codons,
separated by 273 bp, which encode two protein isoforms: LAPTM4B-35
and LAPTM4B-24, with molecular weights of 35 and 24 kDa,
respectively. LAPTM4B-35 is 91 amino acids (N91) longer than
LAPTM4B-24 at the N-terminus. Previous studies have shown that
LAPTM4B-35 is overexpressed in a number of malignant tissues and
has a significant correlation with the prognosis of several types
of cancer, such as hepatocellular (3) and cervical carcinoma (4), breast cancer (5), endometrial carcinoma (6) and ovarian cancer (7). In addition, the LAPTM4B gene has been
demonstrated to promote cell proliferation by regulating cell cycle
control and causing tumorigenesis of NIH3T3 cells, indicating that
it is important in tumorigenesis (8). Furthermore, LAPTM4B-35 promotes the
multidrug resistance of cancer cells (9). By contrast, LAPTM4B-24 is
downregulated in several types of cancer (8,10–12).
Thus, N91 may be a potential candidate for an overexpressed TAA.
However, little is known about the incidence and magnitude of a
pre-existing tumor-specific cellular immune response against N91
protein in patients with cancer. Therefore, the aim of the present
study was to evaluate the potential of N91 protein as a TAA to
induce an antitumor immune response.
Materials and methods
Cell lines, animals and blood
samples
The human tumor cell lines HepG2
(HLA-A*0201+), HeLa, MCF7, Skov3 and T2
(HLA-A*0201+) were maintained in the Department of
Immunology, Cancer Institute, Peking Union Medical College and
Chinese Academy of Medical Sciences (Beijing, China). These human
tumor cells were maintained in RPMI-1640 medium containing 10%
heated-inactivated fetal calf serum, 2 mM L-glutamine, 10 mM HEPES,
penicillin (100 U/ml)-streptomycin (50 μg/ml) solution and 1%
sodium pyruvate solution.
Female C57BL/6 and Balb/c mice were purchased from
the Experimental Animal Institute of Peking Union Medical College
(Beijing, China) and maintained in a specific pathogen-free
environment. The mice were ready for experimental use at six to
eight weeks of age. The Animal Research Ethics Committee of the
Cancer Institute and Hospital, Peking Union Medical College and the
Chinese Academy of Medical Sciences (no. 20120005; Beijing, China)
approved all the protocols involving animals.
Peripheral blood mononuclear cell (PBMC) samples
were obtained from 67 patients with hepatic carcinoma, cervical
carcinoma, breast cancer or ovarian cancer, prior to surgery at the
Cancer Institute and Hospital (Beijing, China) between January 2009
and February 2012. The patient population comprised 41 males and 26
females, with a mean age of 52.64 years (range, 23–81 years). In
addition, 25 blood samples were obtained from healthy donors (19
males and 6 females; median age, 31 years). The Institutional
Ethics Committee of Peking Union Medical College approved the study
prior to its initiation, and written informed consent was provided
by all the participants.
Peptide synthesis and HLA-A*0201
peptide-stabilization assay
The HLA-A*0201-binding peptides in the N91 sequence
were identified using the publicly available peptide-motif scoring
systems (http://www-bimas.cit.nih.gov/molbio/hla_bind/ and
http://www.syfpeithi.de). The potential natural
processing of the peptides by proteasomal cleavage was evaluated
using the Prediction Algorithm for Proteasomal Cleavages website
(http://www.paproc.de). The following three
peptides were identified: GLQARRSTL (N91-1), PLPVPAAAAV (N91-2) and
QARRSTLLKTC (N91-3). The peptides were synthesized by Sangon
Biotech Co., Ltd. (Shanghai, China) and purified (>95%) using
high-performance liquid chromatography. The synthesized peptides
were maintained in dimethylsulfoxide (DMSO), aliquoted at 10 mg/ml
and stored at −80ºC.
Each peptide was examined for the ability to undergo
concentration-dependent binding to a transporter associated with
antigen processing-defective cells (T2) in an HLA-A*0201
stabilization assay (13,14). The T2 cells were incubated at room
temperature overnight, with peptide concentrations of 50–100 μg/ml,
in AIM-V medium (GIBCO® Life technology™, Grand Island,
NY, USA) containing 5 μg/ml β2-microglobulin. Following staining
with HLA-A2-specific monoclonal antibody (mAb; clone BB7.2,
conjugation of PE; Biolegend, San Diego, CA, USA), flow cytometry
was used to assess the stability of HLA-A*0201. A phycoerythrin
(PE)-labeled isotype-matched antibody (mouse IgG2b; Biolegend) was
used as the control. The HLA-A*0201 standard binding peptide, Flu
(GILGFVFTL), was used as a positive control and Msurv33
(LYLKNYRIA), from the H-2d mouse, was used as a negative control
(15). Data were expressed as an
increase in mean fluorescence intensity (MFI) of the cells, with
each peptide compared with the cells without peptide or with a
negative control peptide [FI = (MFI of T2 cells + peptide)/(MFI of
T2 cells with negative peptide)]. FlowJo software (Tree Star, Inc.,
Ashland, OR, USA) was used to analyze the acquired data.
Generation of antigen-specific cytotoxic
T lymphocytes (CTLs)
The glutathione S-transferase (GST)-N91 fusion
protein (supplied by RL Zhou from the Department of Cell Biology,
Peking University Health Science Center, Beijing, China) was
purified by affinity chromatography using GST™ resin (Merck KGaA,
Darmstadt, Germany), in accordance with the manufacturer’s
instructions. SDS-PAGE was performed to analyze the purified
protein from the infected cells. In brief, the separating gel was
12.5% w/v and the stacking gel was 4% w/v. Prior to
electrophoresis, the samples were heated in the presence of sample
buffer at 100ºC for 5 min in a boiling water bath. Each lane
contained 40 μg of protein extraction. Electrophoresis was
performed using a constant voltage of 80 V for 95 min. Proteins
were visualized in gels by staining with 0.025% Coomassie brilliant
blue (R-250, CBB; Sigma, St. Louis, MO, USA) in 50% (v/v) methanol,
10% (v/v) acetic acid. The GST-N91 fusion protein was then digested
with PreScission Protease (GE Healthcare Life Sciences, Piscataway,
NJ, USA) in cleavage buffer. Following the completion of the
digestion, Glutathione Sepharose was added to the sample to remove
the GST protein. The N91 protein was stored at −80ºC for use.
PBMCs were isolated using Ficoll-Paque™ PLUS medium
(GE Healthcare Life Sciences). In brief, the diluted blood sample
was carefully layered on Ficoll-Paque PLUS, then centrifuged at 400
× g for 20–30 min at 18–20ºC. The undisturbed lymphocyte layer was
obtained at the interface. After washing with phosphate buffer
saline, the lymphocytes were used as PBMC sources. The PBMCs and
tumor cell lines were screened for HLA-A*0201 expression by flow
cytometry using an HLA-A2-specific monoclonal antibody (mAb; clone
BB7.2; BioLegend). The genotyping for the HLA-A2 alleles was
performed using reverse transcription-polymerase chain reaction
(RT-PCR) amplification with sequence-specific primers and
sequence-based typing (16). The
HLA-A*0201 subtyping was detected using ThermoScript ™ Reverse
Transcriptase kits (Invitrogen™ Life Technologies, Grand Island,
NY, USA) according to the manufacturer’s instructions. The sequence
of primers and the primer combination were shown in Tables I and II, respectively. The healthy donors and
patients were selected on the basis of HLA-A*0201 antigen
expression (data not shown). Monocyte-derived dendritic cells (DCs)
were generated from PBMCs (HLA-A*0201) as previously described
(17). Mature DCs (mDCs) exhibited
the phenotype CD14low, CD11c+,
CD86high, HLA-DRhigh and
MHC-Ihigh. mDCs were pulsed with 50 or 100 μg/ml control
peptide or 20 μg/ml N91 protein in serum-free AIM-V medium
overnight.
 | Table IPrimers for the detection of the
HLA-A*0201. |
Table I
Primers for the detection of the
HLA-A*0201.
| Primer | Sequence (5′-3′) |
|---|
| Genomic primers | |
| Sense-primer | |
| AL#37 | CCT CGT CCC CAG GCT
CT |
|
Antisense-primer | |
| AL#AW | TGG CCC CTG GTA CCC
GT |
| Nested primers | |
| Sense-primer | |
| AL#22 | CAC TCC ATG AGG TAT
TTC TT |
| AL#14 | AGG CCC ACT CAC AGA
CTC |
| AL#3 | GAC GGG GAG ACA CGG
AAA |
|
Antisense-primer | |
| AL#Q | CTC CAG GTA GGC TCT
CAA |
| AL#BG | CGT CGC AGC CAT ACA
TCC |
| AL#BF | CCC CAC GTC GCA GCC
AT |
| AL#V | GAG CCA CTC CAC GCA
CGT |
| Table IIReaction combinations for the
detection of the HLA-A*0201. |
Table II
Reaction combinations for the
detection of the HLA-A*0201.
| Primer
combination | Product size
(bp) |
|---|
| Step1 |
| AL#37, AL#AW | 812 |
| Step2 |
| AL#22, AL#Q | 718 |
| AL#22, AL#BG | 542 |
| AL#22, AL#BF | 547 |
| AL#14, AL#V | 543 |
| AL#3, AL#V | 565 |
To induce CTLs in vitro, the PBMCs
(2×106/ml) were suspended in serum-free medium and
divided into two fractions. One of the fractions was cultured with
a pool of the three N91 peptide-pulsed DCs, while the other
fraction was incubated with N91 protein-pulsed DCs
(2×105/ml) in 6-well plate (1 ml per well). The CTLs
were generated by two cycles of stimulation with peptide or N91
protein-loaded DCs every seven days (DC:PBMC = 1:10) (18). The CTL functions were analyzed
using interferon-γ (IFN-γ) enzyme-linked immunosorbent spot
(ELISPOT) assay kits (BD-Pharmingen, San Jose, CA, USA), in
accordance with the manufacturer’s instructions. Typically, a fixed
number of various target and effector cells (5×104 cells
per well, effector to HepG2 target ratio of 40:1) were cultured in
replicate wells overnight, and the spots were quantified using an
immunospot reader (Cellular Technology Limited, Shaker Heights, OH,
USA). The results were presented as the number of IFN-γ-producing
cells per 5×104 cells.
Tissue collection, protein extracts and
western blot analysis
The tissue samples from the C57BL/6 mice were
flash-frozen in liquid nitrogen immediately subsequent to
collection and lysed in radioimmunoprecipitation assay (RIPA) lysis
buffer. PBMCs or splenocytes from the C57BL/6 and Balb/c mice were
activated with phytohemagglutinin (5 μg/ml) or concanavalin-A (4
μg/ml) for 72 h, respectively. The cells were then harvested and
lysed in prechilled RIPA lysis buffer. Following centrifugation at
10,000 × g for 15 min at 4ºC, the supernatant was collected for
western blotting. Protein concentration was analyzed by monitoring
the visible spectrophotometer absorbance at 595 nm. For western
blotting, the protein was separated using 12.5% SDS-PAGE, and the
proteins were transferred onto Immobilon-FL polyvinylidene
difluoride (PVDF) membranes (Millipore, Billerica, MA, USA) using a
wet-transfer apparatus (Bio-Rad, Hercules, CA, USA). The membranes
were subsequently blocked by incubation in 5% non-fat dry milk,
washed and incubated with specific antibody [rabbit immunoglobulin
G (IgG), supplied by RL Zhou], followed by secondary reagents
[anti-rabbit IgG labeled with horseradish peroxidase (HRP); Cell
Signaling Technology, Danvers, MA, USA]. The membranes were
visualized using an enhanced chemiluminescence (ECL) western
blotting system (ECL western blotting system, GE Healthcare Life
Sciences). As an internal loading control, β-actin was probed with
a commercial antibody (Cell Signaling Technology, Inc.).
Statistical analysis
Data were expressed as the mean ± standard error of
the mean (S.E.). Differences between groups were evaluated using
one-way analysis of variance (ANOVA). P<0.05 was considered to
indicate a statistically significant difference. Statistical
analyses were performed using the commercially available software
SPSS 16.0 (SPSS, Inc., Chicago, IL, USA).
Results
Stabilization of HLA-A*0201 on a T2 cell
line
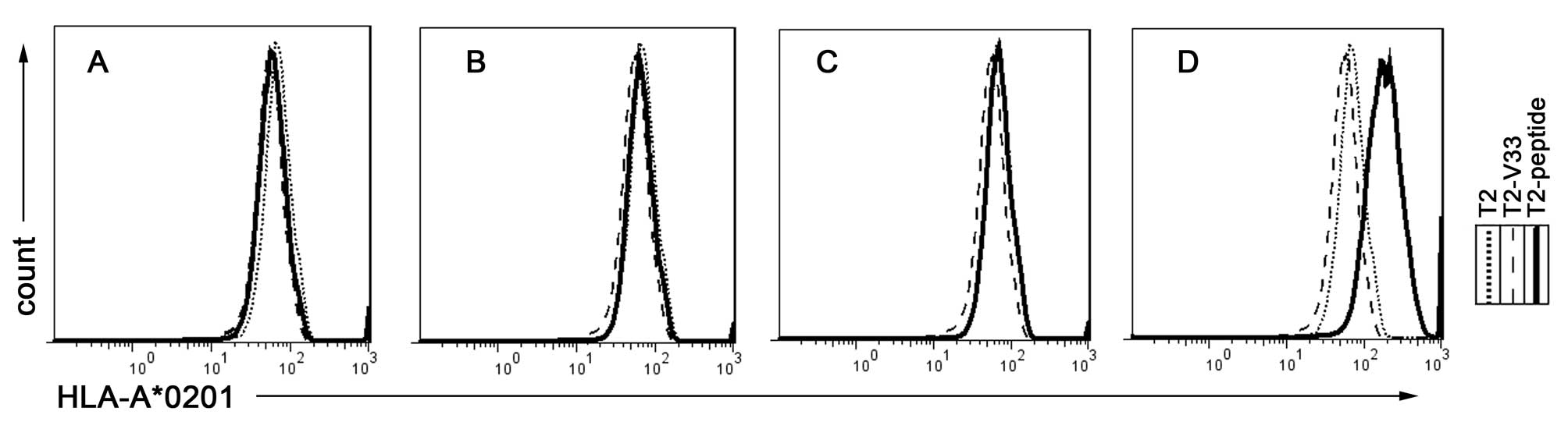
To assess whether synthetic peptides were able to
stabilize the expression of the HLA-A*0201 molecule on the T2 cell
surface, peptide-induced HLA-A*0201 upregulation on the T2 cells
was examined according to a previously described protocol (19). When three different peptides of the
N91 sequence (Table III) were
co-cultured with the T2 cells, the peptides weakly enhanced the
surface expression of HLA-A*0201 (Fig.
1A–C). However, pulsing the cells with the positive control
peptide (Flu) notably stabilized the HLA-A*0201 expression on the
surface of the T2 cells (Fig. 1D).
The binding data are shown in Table
I: All three examined peptides were observed to weakly
stabilize HLA-A*0201. In general, the MHC stabilization by these
peptides was more consistent with the calculated score based on the
half-life of dissociation of the peptide from the HLA molecule than
the binding affinity as calculated by the SYFPEITHI epitopes
prediction.
| Table IIIHLA-A*0201 epitope affinity assay. |
Table III
HLA-A*0201 epitope affinity assay.
| Peptide | Amino acid
sequence | Fluorescence
intensity |
|---|
| N91-1 | GLQARRSTL | 0.98 |
| N91-2 | PLPVPAAAAV | 0.90 |
| N91-3 | QARRSTLLKTC | 0.90 |
| Influenza (Flu) | GILGFVFVFTL | 2.70 |
| Msurv33 (V33) | LYLKNYRIA | 0.90 |
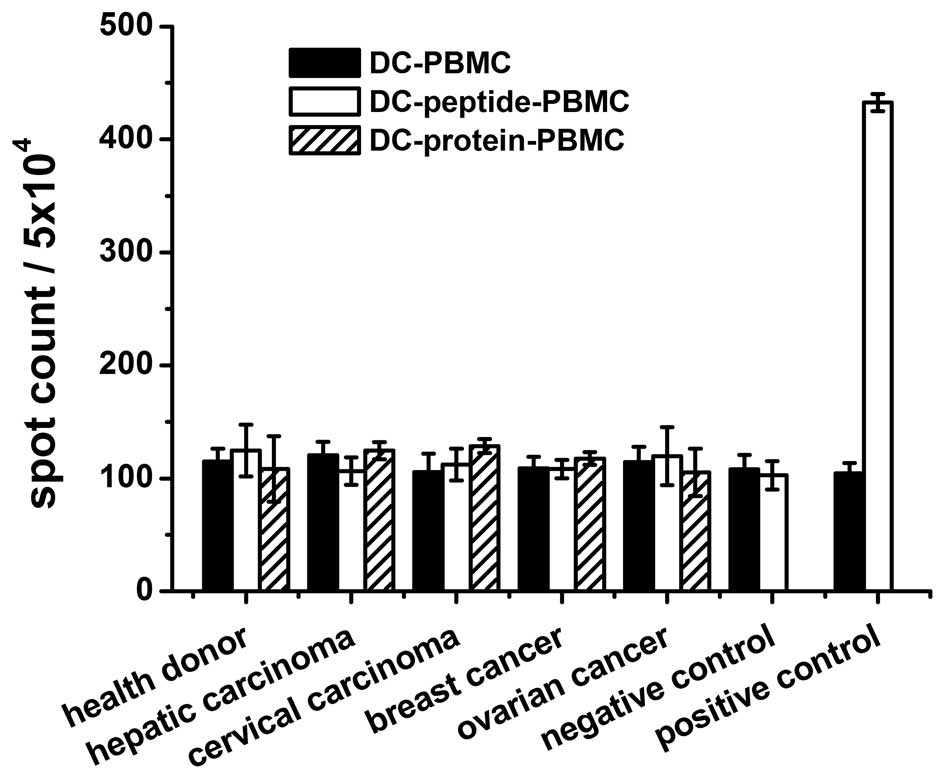
CTL responses against peptides or N91
protein-loaded targets
It was assessed whether the peptides (N91-1 one of
the 3 peptides tested, amino acid sequence is ‘GLQARRSTL’) or N91
protein were able to bind to HLA-A*0201+ DCs and induce
a CTL response against the target cells (HepG2 and
HLA-A*0201+) loaded with N91 protein. The responses of
PBMCs to N91 peptide or protein were examined in 67
HLA-A*0201+ patients, while the frequency of the T-cell
response to N91 peptide or protein, positive Flu peptide and the
negative control, V33 peptide, was evaluated in healthy donors. The
ELISPOT assay was considered to be the most sensitive and feasible
screening test for the N91-specific T-cell assay. As shown in
Fig. 2, the activation of CTLs was
undetectable or negligible. No reactivity was observed for the N91
peptide or protein in the patients or healthy donors. The
B16F10-N91 tumor-bearing mice experiments demonstrated that the N91
specific immune response was also negligible (data not shown).
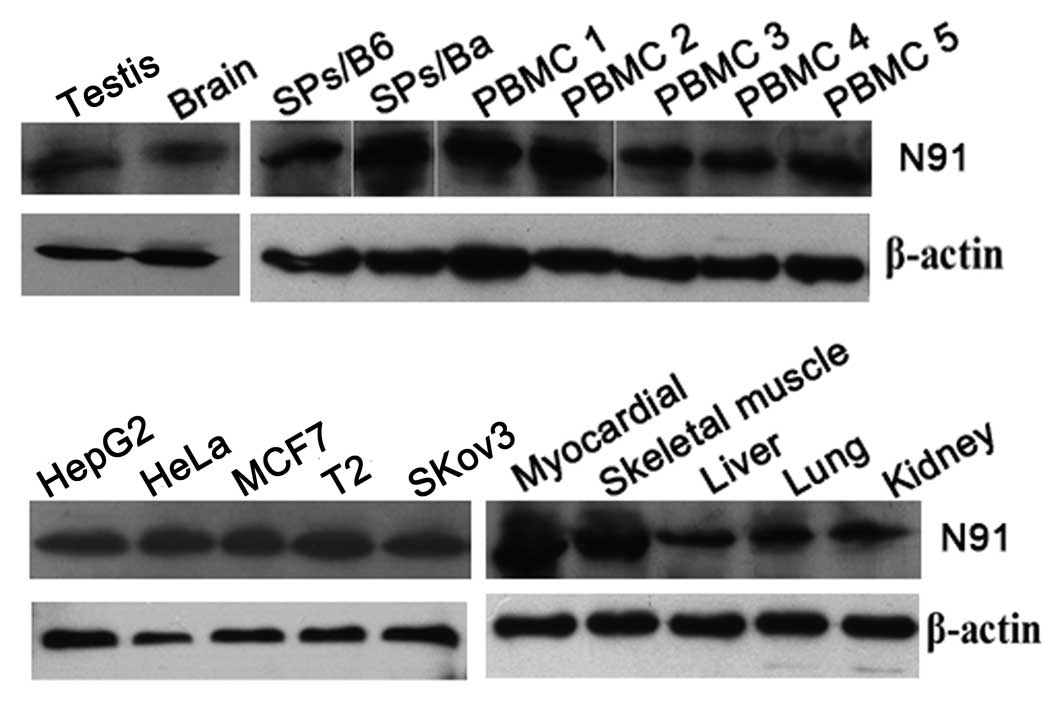
Expression level of the N91 protein in
normal and cancer cells
The previously described experiments showed that
CTLs against N91 protein were not able to be induced in
vitro. To elucidate the mechanism underlying this phenomenon,
the expression levels of N91 proteins from normal tissues or cells
were examined using western blot analysis. Human LAPTM4B shares 92%
homology with mouse LAPTM4B at the amino acid level, suggesting
that LAPTM4B has been highly conserved within vertebrate species
(8). Thus, the expression level of
the N91 protein was assessed in mouse tissues. As shown in Fig. 3, N91 protein expression was
detected by western blotting in certain tumor cell lines and normal
tissues, including the lysates of activated PBMCs from normal
healthy donors, lysates of the liver, kidney, brain, testis,
skeletal muscle and myocardium, and activated splenocytes from
naïve C57BL/6 and Balb/c mice. No significant differences were
identified between the expression levels of N91 protein in the
tumor cell lines and the normal tissue. Thus, it was suggested that
the widespread expression of the N91 protein may be responsible for
the weak MHC class I processing and epitope presentation.
Discussion
The majority of cancer antigens are ‘self-antigens’,
expressed on normal cells. Immunogenic peptides derived from these
TAAs have been used in therapeutic vaccines (20,21).
Synthetic peptides from the predicted high-binding category are
loaded on HLA-matched DCs or other antigen-presenting cells, and
used as stimulators in CTL induction. The LAPTM4B-35 protein is
significantly upregulated in several types of cancer (8), indicating that it may be a candidate
tumor antigenic epitope. According to the SYFPEITHI analysis of the
N91 protein sequence, three different peptides were synthesized.
However, the results demonstrated that it was not possible to
induce an immune response using peptide-pulsed DCs co-cultured with
PBMCs in vitro, as examined using the IFN-γ assay. Previous
studies with other cancer-specific peptides have demonstrated that
CTL responses are able to be induced by peptides with various MHC
binding affinities (22,23). Furthermore, it has been shown that
tolerance to a self-peptide is most efficiently established by
those self-peptides with a high-binding affinity for MHC (24). Alternatively, a lower binding
affinity may induce an antitumor immune response to some degree.
Thus, a N91 protein-pulsed DCs experiment was performed to
investigate this. However, there no significant differences were
identified between the DC-protein-PBMC and DC-PBMC groups. The high
expression of the N91 protein in certain normal tissues and
activated PBMCs, as well as in malignant cells, may have been
responsible for the negative result. The results suggested that the
overexpression of the N91 protein may not be a candidate for a
TAA.
Certain aspects of currently available technologies
inhibit the identification of novel tumor antigens. Differential
genomics and proteomics approaches identify over- and
underexpressed proteins. However, these methods are unable to
identify very low-abundance proteins that are often processed and
presented by MHC class I molecules as the true rejection targets
for T cells. Furthermore, the level of protein expression does not
always correlate with MHC processing and presentation in cancer
(25). In addition, a study of
tumor immunology suggested that tumor development is associated
with the induction of tolerance to tumor antigens, leading to
immunosuppression, which prevents an autologous adoptive
immunotherapy for patients with refractory metastatic solid tumors
(26). In patients with cancer,
spontaneous antitumor immune responses appear to be too weak to
cause the spontaneous rejection of the tumors, since the cancer
continues to develop (27). A
major concern for the use of tumor-derived antigens is the
possibility that the epitope may activate autoimmune disease.
Furthermore, many of these peptides are not tumor-restricted. This
may underlie the mechanism whereby the overexpression of the N91
protein is not recognized by DCs as a tumor antigen.
Therefore, the identification of novel TAAs remains
one of the major aims for the design of more effective
immunological treatments for cancer. Ideal targets for
immunotherapy are gene products that are silenced in normal tissues
and only overexpressed in cancer cells, and that are directly
involved in tumor survival and progression. The weak immunogenicity
of N91 protein may be explained by its increased expression in
certain normal tissues, as well as malignant cells. Thus, it
appears to be a weak candidate for a TAA.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 30872373). The authors would
like to thank all the individuals who participated in this
study.
References
|
1
|
Tsang KY, Zaremba S, Nieroda CA, Zhu MZ,
Hamilton JM and Schlom J: Generation of human cytotoxic T cells
specific for human carcinoembryonic antigen epitopes from patients
immunized with recombinant vaccinia-CEA vaccine. J Natl Cancer
Inst. 87:982–990. 1995. View Article : Google Scholar
|
|
2
|
Palermo B, Del Bello D, Sottini A, et al:
Dacarbazine treatment before peptide vaccination enlarges T-cell
repertoire diversity of melan-a-specific, tumor-reactive CTL in
melanoma patients. Cancer Res. 70:7084–7092. 2010. View Article : Google Scholar
|
|
3
|
Toes RE, Blom RJ, van der Voort E,
Offringa R, Melief CJ and Kast WM: Protective antitumor immunity
induced by immunization with completely allogeneic tumor cells.
Cancer Res. 56:3782–3787. 1996.PubMed/NCBI
|
|
4
|
Meng F, Song H, Luo C, et al: Correlation
of LAPTM4B polymorphisms with cervical carcinoma. Cancer.
117:2652–2658. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xiao M, Jia S, Wang H, Wang J, Huang Y and
Li Z: Overexpression of LAPTM4B: an independent prognostic marker
in breast cancer. J Cancer Res Clin Oncol. 139:661–667. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Meng F, Li H, Zhou R, Luo C, Hu Y and Lou
G: LAPTM4B gene polymorphism and endometrial carcinoma risk and
prognosis. Biomarkers. 18:136–143. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xu Y, Liu Y, Zhou R, et al: LAPTM4B
polymorphisms is associated with ovarian cancer susceptibility and
its prognosis. Jpn J Clin Oncol. 42:413–419. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shao GZ, Zhou RL, Zhang QY, et al:
Molecular cloning and characterization of LAPTM4B, a novel gene
upregulated in hepatocellular carcinoma. Oncogene. 22:5060–5069.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu XS, Dyer J, Leggatt GR, et al:
Overcoming original antigenic sin to generate new CD8 T cell
IFN-gamma responses in an antigen-experienced host. J Immunol.
177:2873–2879. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang H, Zhai G, Ji X, Xiong F, Su J and
McNutt MA: LAPTM4B allele *2 is a marker of poor prognosis
following hepatic tumor resection for hepatocellular carcinoma.
PLoS One. 7:e349842012.
|
|
11
|
Zhai G, Yan K, Ji X, et al: LAPTM4B allele
*2 is a marker of poor prognosis for gallbladder carcinoma. PLoS
One. 7:e452902012.
|
|
12
|
Zhou L, He XD, Chen J, et al:
Overexpression of LAPTM4B-35 closely correlated with
clinicopathological features and post-resectional survival of
gallbladder carcinoma. Eur J Cancer. 43:809–815. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kirk CJ, Hartigan-O’Connor D, Nickoloff
BJ, et al: T cell-dependent antitumor immunity mediated by
secondary lymphoid tissue chemokine: augmentation of dendritic
cell-based immunotherapy. Cancer Res. 61:2062–2070. 2001.PubMed/NCBI
|
|
14
|
Wang W, Epler J, Salazar LG and Riddell
SR: Recognition of breast cancer cells by CD8+ cytotoxic T-cell
clones specific for NY-BR-1. Cancer Res. 66:6826–6833. 2006.
|
|
15
|
Parker KC, Bednarek MA and Coligan JE:
Scheme for ranking potential HLA-A2 binding peptides based on
independent binding of individual peptide side-chains. J Immunol.
152:163–175. 1994.PubMed/NCBI
|
|
16
|
Gatz SA, Pohla H and Schendel DJ: A
PCR-SSP method to specifically select HLA-A*0201 individuals for
immunotherapeutic studies. Tissue Antigens. 55:532–547.
2000.PubMed/NCBI
|
|
17
|
Neumann F, Wagner C, Preuss KD, et al:
Identification of an epitope derived from the cancer testis antigen
HOM-TES-14/SCP1 and presented by dendritic cells to circulating
CD4+ T cells. Blood. 106:3105–3113. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Neumann F, Wagner C, Stevanovic S, et al:
Identification of an HLA-DR-restricted peptide epitope with a
promiscuous binding pattern derived from the cancer testis antigen
HOM-MEL-40/SSX2. Int J Cancer. 112:661–668. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nolte A, Scheffold C, Slotty J, et al:
Generation of melanoma-specific cytotoxic T lymphocytes for
allogeneic immunotherapy. J Immunother. 26:257–269. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Boon T: Tumor antigens recognized by
cytolytic T lymphocytes: present perspectives for specific
immunotherapy. Int J Cancer. 54:177–180. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rammensee H, Bachmann J, Emmerich NP,
Bachor OA and Stevanovic S: SYFPEITHI: database for MHC ligands and
peptide motifs. Immunogenetics. 50:213–219. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Barnea E, Beer I, Patoka R, et al:
Analysis of endogenous peptides bound by soluble MHC class I
molecules: a novel approach for identifying tumor-specific
antigens. Eur J Immunol. 32:213–222. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ramakrishna V, Ross MM, Petersson M, et
al: Naturally occurring peptides associated with HLA-A2 in ovarian
cancer cell lines identified by mass spectrometry are targets of
HLA-A2-restricted cytotoxic T cells. Int Immunol. 15:751–763. 2003.
View Article : Google Scholar
|
|
24
|
Fairchild PJ and Wraith DC: Lowering the
tone: mechanisms of immunodominance among epitopes with low
affinity for MHC. Immunol Today. 17:80–85. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shastri N, Schwab S and Serwold T:
Producing nature’s gene-chips: the generation of peptides for
display by MHC class I molecules. Annu Rev Immunol. 20:463–493.
2002.
|
|
26
|
Ren XB, Yu JP, Cao S, et al: Antitumor
effect of large doses IL-2-activated HLA haploidentical peripheral
blood stem cells on refractory metastatic solid tumor treatment.
Cancer Biother Radiopharm. 22:223–234. 2007. View Article : Google Scholar
|
|
27
|
Smyth MJ, Godfrey DI and Trapani JA: A
fresh look at tumor immunosurveillance and immunotherapy. Nat
Immunol. 2:293–299. 2001. View
Article : Google Scholar : PubMed/NCBI
|