Introduction
Hepatocellular carcinoma (HCC) is one of the most
common tumors worldwide, and its incidence is unlikely to be
reduced in the near future, despite the developments in surgical
treatment. In China and Southeast Asia, the incidence of HCC is
high, with HCC occurring most frequently following the development
of chronic liver disease resulting from infection with hepatitis
virus (1). Therefore, it is
important to elucidate the molecular mechanism of HCC
carcinogenesis and to develop specific measures for the prevention
of HCC.
Fragile histidine triad (FHIT) protein belongs to
the family of the evolutionary conserved histidine triad (HIT)
proteins, which consists of at least three subfamilies: FHIT,
histidine triad nucleotide binding protein 1 (HINT1) and
galactose-1-phosphate uridylyltransferase (GALT) (2). Our previous study showed that HINT1
functions as a tumor-suppressor gene in human hepatoma cell lines
(3). FHIT is located at human
chromosome 3p14.2, and encodes a transcript of 1.1 kb (4). It encompasses the FRA3B fragile site
and a genomic locus that is frequently involved in cytogenetic
abnormalities, genomic rearrangement and allelic loss in tumors
(5). The aberrant methylation of
normally unmethylated CpG islands, located in the 5′ promoter
region of genes, is associated with the transcriptional
inactivation of a number of genes in human cancer and may serve as
an alternative to mutational inactivation. The hyper-methylation of
the 5′ promoter region of the FHIT tumor-suppressor gene has been
observed in human cell lines of leukemia, as well as in breast,
pancreatic and esophageal cancer (6–9).
However, the methylation status of FHIT in human hepatoma cells
requires further investigation.
The Wnt/β-catenin/transcription factor 4 (TCF4)
signaling pathway is important in the survival, proliferation and
differentiation of cells (10).
Our previous study showed that in HepG2 human hepatoma cells, the
HIT family member HINT1 binds to the Pontin/Reptin/β-catenin/TCF4
complex, and thus deactivates TCF4 (3). Mutations in the adenomatous polyposis
coli (APC) gene have not been observed in experimental or human
hepatoma. The increased expression of β-catenin has been correlated
with a poor prognosis in patients with hepatoma. Following the
activation of the Wnt/β-catenin/TCF4 pathway, cyclin D1 is induced
to form an active enzyme complex with cyclin-dependent kinase 4/6
(CDK4/6), which phosphorylates and activates CDK and leads to the
G1/S transition (11). As a
result, cell proliferation and tumor genesis are induced. The
activation of the Wnt/β-catenin/TCF4 pathway and the overexpression
of cyclin D1 in human HCC cells have been studied previously
(12,13).
The purpose of the present study was to examine the
expression and function of FHIT in human hepatoma cells. These
cells were the subject of the study due to the unusual role of
β-catenin in hepatoma cells and the global prevalence of
hepatoma.
Materials and methods
Cell lines and cell culture
The human hepatoma cell lines HepG2, Hep3B and Huh7
were obtained from the American Type Culture Collection (Manassas,
VA, USA). The cell lines were maintained in a DF10 medium
containing Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen
Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal
bovine serum (FBS; Invitrogen Life Technologies), and were
incubated in a 100% humidified incubator at 37°C with 5%
CO2.
Protein extraction and western blot
analysis
The cells were cultured and treated as described
above. The cells were collected and resuspended in 10 ml of lysis
buffer (25 mM Tris-HCl, pH 7.5, 20 mM NaCl, 1 mM EDTA, 20% (v:v)
glycerol, 1% (v:v)Triton X-100, 2X protease inhibitor mixture for
proteins, respectively. After sonication (4×20 sec, 1 min between
cycles), the lysates were centrifuged at 12,000 × g at 40°C for 20
min. The proteins were separated using SDS-PAGE with 7.5–12.5%
polyacrylamide gels at 4°C for 2 h and subsequently blotted with
the indicated antibodies at 4°C overnight. The primary antibodies
included anti-FHIT, anti-hemagglutinin (HA)-Tag, anti-cyclin D1 and
anti-β-actin (all from Sigma, St. Louis, MO, USA). Antimouse and
antirabbit IgA (Amersham Biosciences, New York, NY, USA) antibodies
were used as the secondary antibodies. Each membrane was developed
with an enhanced chemiluminescence system (Amersham Biosciences).
The intensities of specific protein bands were quantified with NIH
Image software version 1.62 (NIH, Bethesda, MD, USA), corrected for
the intensity of the respective β-actin band.
Methylation-specific polymerase chain
reaction (PCR)
Total cell DNA was isolated from the cultured cells
using a QIAamp DNA Mini kit (Qiagen N.V., Venlo, The Netherlands).
The genomic DNA was modified as described previously (14) using a DNA bisulfite modification
kit (Chemicon, Temecula, CA, USA). The methylated CpG dinucleotides
within a CpG island in the promoter region of FHIT were detected
using the following specific PCR primer sets: methylated FHIT
forward, 5-TTGGGGCGCGGGTTTGGGTTTTTACGC-3 and reverse,
5-CGTAAACGACGCCGACCCCACTA-3; and unmethylated FHIT forward,
5-TTGGGGTGTGGGTTT GGGTTTTTATG-3 and reverse,
5-CATAAACAACACCAACCCCACTA-3 (15).
Each PCR mixture system contained 10X PCR buffer (Qiagen N.V.),
deoxynucleotide triphosphates (1.25 mmol/l), primers (0.6 mmol/l),
1 unit HotStarTaq® (Qiagen N.V.), and bisulfite-modified
DNA (100 ng). PCR was performed in a programmable thermal
controller (MJ Research, Inc., Quebec, Canada) under the following
conditions: Preheating at 94°C for 3 min, 94°C for 30 sec, 62°C for
30 sec and 72°C for 30 sec for 38 cycles and a final extension at
72°C for 7 min. The PCR products were analyzed on a 2% agarose
gel.
5-Aza-2′-deoxycytidine (5-azadc)
treatment and FHIT expression
The cells were treated with a solution or with
5-azadc (1 μmol/l; Sigma) for 96 h. Following the extraction of
total proteins from the cells, the expression levels of FHIT
protein were examined using western blotting.
Plasmid construction and gene
transduction
Full-length human FHIT cDNA was amplified from
HCT116 cells with a one-step reverse transcription (RT)-PCR
procedure, using the primers 5-CCAATGGATCCATGTCGTTCAGATTTGG-3
(forward) and 5-CCAATCTCGAGTCACTGAAAGTAG ACC-3 (reverse) (16). The PCR products were then cloned
into the BamHI/XhoI-treated plasmid pHANE, which
contained the NH2-terminal HA epitope tag (YPYDVPDYA)
(14). The sequence of the
pHA-FHIT construct was confirmed using DNA sequencing.
Cell proliferation assays
Colony formation
HepG2 cells were transfected with the pHA-FHIT
plasmid and the empty control plasmid pcDNA3. The cells were
subsequently replated into 100-mm dishes at 1×105 cells
per dish, and were cultured in the presence of 0.8 mg/ml G418 for
three weeks. The colonies were then fixed with formalin, stained
with Giemsa and counted.
Growth curves
HepG2 cells were transfected with the PcDNA3
(Invitrogen Life Technologies) and HA-FHIT (constructed by
Professor Lin Wang) plasmids and counted every day for eight days.
The number of viable cells per well was determined each day. Each
assay was performed in triplicate.
Luciferase reporter assays
The HepG2 cells were plated at 1×105
cells per 35-mm-diameter plate 18 h prior to transfection. The
cells were subsequently transfected with the p-1745-CD1-LUC
luciferase reporter plasmid (400 ng/well) (and 200 ng/well
p-cytomegalovirus (CMV)-β-galactosidase reporter plasmid, with or
without cotransfection with HA-FHIT plasmid (400, 300, 200 or 100
ng) DNA. The p-1745-CD1-LUC, CMV-β-galactosidase and wild-type
β-catenin plasmids were provided by Kathryn Calame (Columbia
University, New York, NY, USA). At 36 h post-transfection, cell
extracts were prepared, and each sample was assayed in triplicate,
using a Luciferase assay system (Promega Corporation, Madison, WI,
USA). Luciferase activities were normalized to β-galactosidase
activities to correct for the differences in transfection
efficiency. Significant differences (P<0.05) with respect to the
corresponding control assay are indicated by an asterisk (*).
Statistical analysis
All data are presented as the mean ± standard
deviation (SD). P<0.05 was considered to indicate a
statistically significant difference. The statistical analysis was
performed using SPSS version 11.0 statistical software (SPSS, Inc.,
Chicago, IL, USA).
Results
Expression of FHIT in human hepatoma
cells
Western blotting indicated that FHIT protein was
expressed at a low level in HepG2 and Hep3B cells; however, it was
expressed at a relatively high level in Huh7 cells (Fig. 1A). Similar results were obtained in
a repeated study.
Methylation status of FHIT in hepatoma
cells
The expression levels of certain specific
tumor-suppressor genes may be inhibited in cancer cells via the
methylation of cytidine residues in the corresponding promoter
regions of these genes, or via other modifications that alter the
structure of chromatin. Therefore, the methylation status of a
CpG-rich region in the promoter region of FHIT was examined using
bisulfite-treated DNA samples obtained from the three cell lines,
with sets of methylation-specific primers. Partial methylation of
the DNA was observed in the HepG2 and Hep3B cell DNA samples. No
methylation of the DNA was found in Huh7 cells (Fig. 1B).
The results suggest that the relatively low levels
of FHIT in HepG2 and Hep3B cells may have been at least partially
due to the methylation of the promoter. Therefore, the three cell
lines were treated with 1 μM 5-azadc, the DNA demethylation agent,
for 96 h and then the protein expression levels of FHIT were
examined using western blotting. In the HepG2 and Hep3B cells,
treatment with 5-azadc led to a ~2-fold increase in the level of
FHIT expression (Fig. 1C). By
contrast, similar treatment with 5-azadc did not increase the FHIT
expression levels in the Huh7 cells (Fig. 1C). Therefore, the results in
Fig. 1 demonstrated that the
relatively low-level expression of FHIT in HepG2 and Hep3B cells
was at least in part due to the partial methylation of the promoter
region of this gene.
Increased expression of FHIT inhibits the
growth of HepG2 cells
A plasmid pHA-FHIT was constructed, which contained
full-length FHIT open reading frame and an NH2-terminal
HA epitope tag (YPYDVPDYA) (14).
The constructed plasmid, pHA-FHIT, was confirmed by gene sequencing
(data not shown). Subsequently, the plasmid encoding HA-FHIT or an
empty control plasmid was used to transfect the HepG2 cell line.
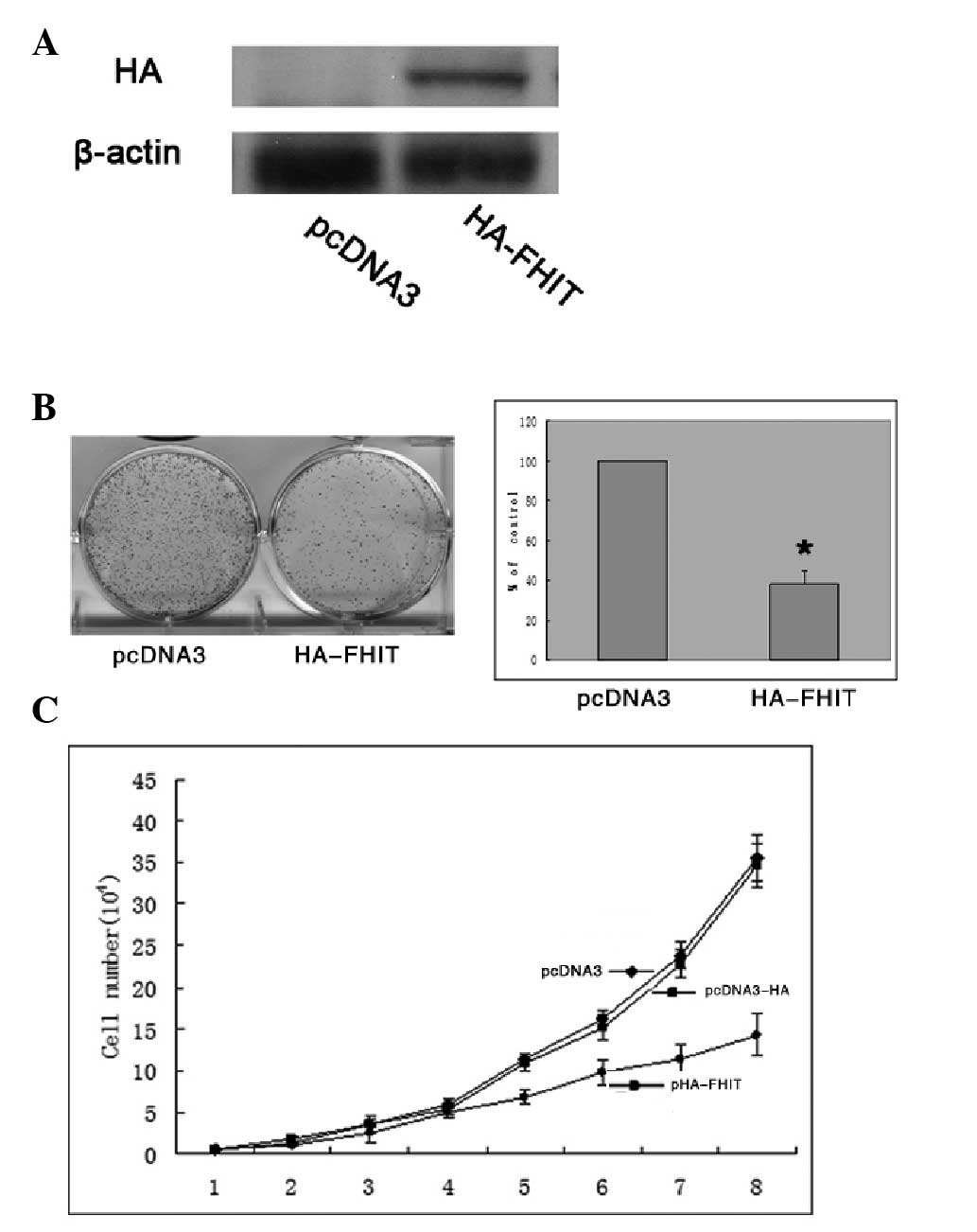
Transfection with HA-FHIT plasmid resulted in overexpression levels
of the associated HA proteins (Fig.
2A). The overexpression of HA-FHIT inhibited colony formation
by ~60% (P<0.05) compared with that of the control
plasmid-transfected cells (Fig.
2B). In addition, cell proliferation assays were performed over
a period of eight days, and cell proliferation was ultimately
inhibited by ~50% in the HA-FHIT-transfected cells compared with
the proliferation of the control plasmid-transfected cells
(Fig. 2C).
FHIT inhibits cyclin D1 expression in
HepG2 cells
Cyclin D1 is a protein involved in the cell cycle
that is affected in the G1 phase of cell cycle. Western blotting of
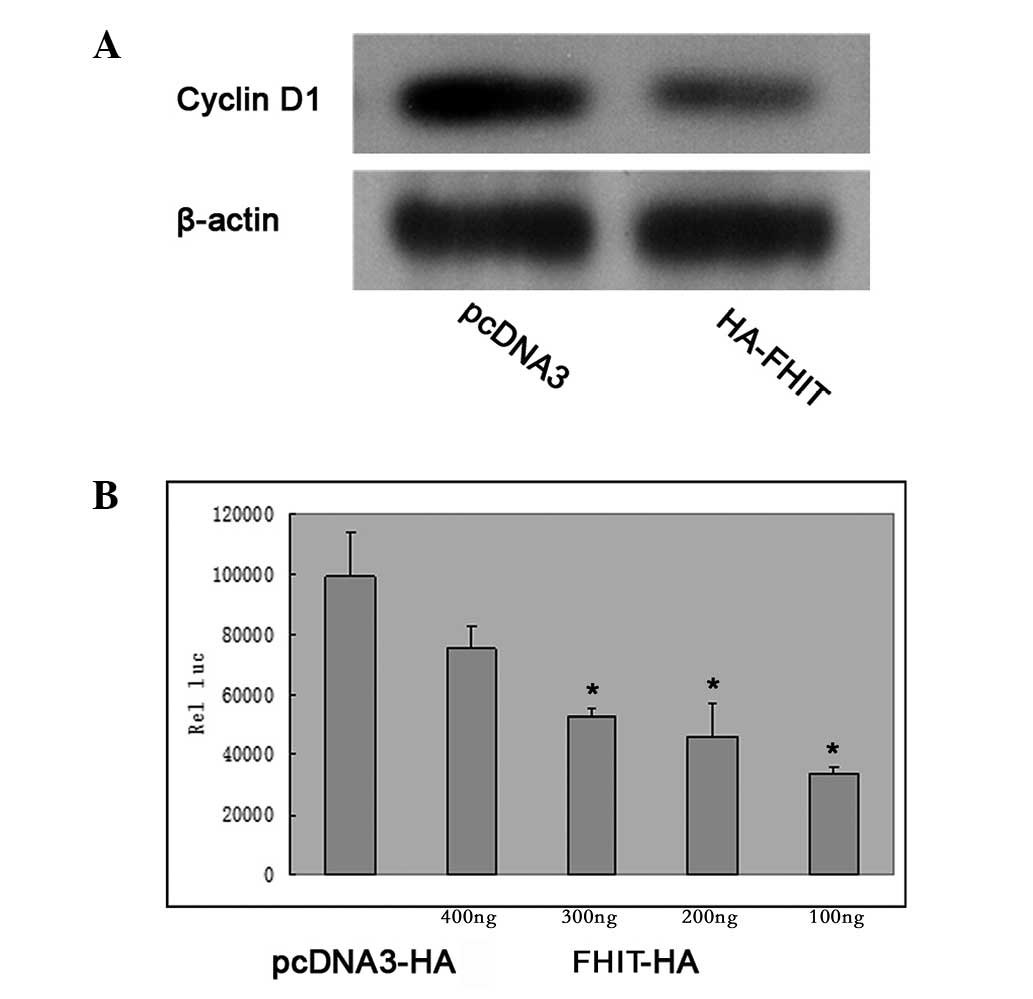
HepG2 cells transfected with HA-FHIT demonstrated that the
overexpression of HA-FHIT inhibited the expression of cyclin D1 in
the cells (Fig. 3A). In HepG2
cells which were transfected with a full-length cyclin D1
promoter-luciferase reporter (p-1745-CD1-LUC), cotransfection with
increasing quantities of FHIT plasmid DNA caused a
concentration-dependent inhibition of the transcriptional activity
of the cyclin D1 promoter (Fig.
3B).
FHIT inhibits the activity of cyclin D1
transcription factor through the β-catenin/TCF4 signaling
pathway
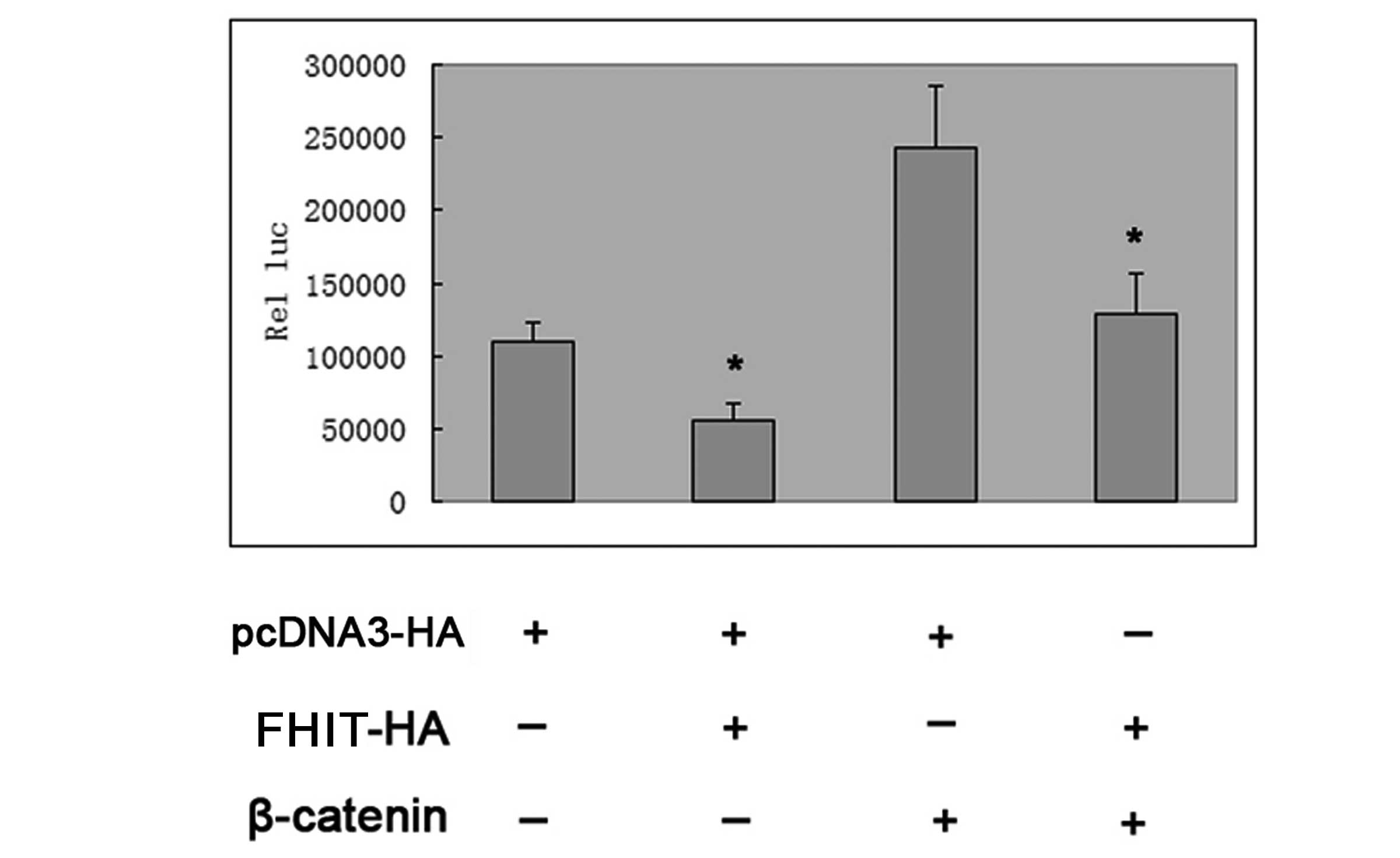
Transfection with plasmid DNA encoding wild-type
β-catenin markedly activated the p-1745-CD1-LUC reporter in HepG2
cells; however, this activation was largely inhibited by additional
cotransfection with FHIT plasmid DNA (Fig. 4). In combination, the results
indicated that FHIT was capable of inhibiting the expression of
cyclin D1 in HepG2 cells.
Discussion
In view of the prevalence of HCC in Asia and Africa
and the rising incidence and rates of mortality of HCC in the
United States (1), the aim of the
present study was to examine a number of molecular aspects of the
function of FHIT in human hepatoma cells. A low-level expression of
FHIT was observed in HepG2 and Hep3B cell lines compared with that
of the Huh7 cell line (Fig. 1A).
One key mechanism for the loss of function of tumor-suppressor
genes in cancer involves gene silencing, mediated by
hyper-methylation or aberrant promoter DNA. Previous studies
demonstrated that the FHIT 5′ promoter region was partially
methylated in certain human cancer cell lines (7,17–19).
In the present study, methylation-specific PCR was used to show
that the FHIT promoter was partially hyper-methylated in HepG2 and
Hep3B cells (Fig. 1B), and that
treatment with 5-azadc increased the expression of FHIT in these
cells. Thus, it was concluded that FHIT was expressed at a low
level in HepG2 and Hep3B cells, which was at least partially due to
the hemi-methylation of the FHIT promoter in these cell lines.
Previous studies have demonstrated that the
overexpression of FHIT, following transfection or infection of the
cells with exogenous FHIT, may significantly inhibit cell growth
and induce apoptosis in leukemia, as well as breast and esophageal
cancer cell lines (20–23). In order to directly show that FHIT
acts as a tumor-suppressor gene in hepatoma cells, the wild-type
FHIT was generated and incorporated into an N′-terminal HA-tagged
FHIT plasmid. Following transient transduction, FHIT-overexpressing
cells were obtained. Colony formation and growth curve assays
showed that the overexpression of exogenous FHIT significantly
inhibited the growth of HepG2 cells (Fig. 2).
The HIT family is characterized by a common
His-X-His-X-His-XX motif, in which X represents a hydrophobic amino
acid (24). In our previous study,
another HIT family member, HINT1, was demonstrated to bind to the
Pontin/Reptin/β-catenin/TCF4 complex, inhibiting the activity of
TCF4 and, thus, significantly inhibiting the expression of its
direct substrate, cyclin D1. This activity of HINT1 was significant
with regard to the cell cycle and cell proliferation (3). Therefore, it was of interest to
investigate whether FHIT was also important in the β-catenin/TCF4
signaling pathway. When HepG2 cells were transfected with a
full-length cyclin D1 promoter-luciferase reporter
(p-1745-CD1-LUC), and cotransfected with increasing quantities of
FHIT plasmid DNA, a concentration-dependent inhibition of the
transcriptional activity of the cyclin D1 promoter was observed
(Fig. 3B). Transfection with
plasmid DNA encoding wild-type β-catenin markedly stimulated the
activity of this reporter in HepG2 cells; however, this stimulatory
activity was largely inhibited by additional cotransfection with
FHIT plasmid DNA (Fig. 4). In
order to directly examine the effects of FHIT on the expression of
the endogenous cyclin D1 gene in HepG2 cells, these cells were
transfected with pHA-FHIT, which encoded the FHIT cDNA sequence,
(Fig. 2A). It was observed that
overexpression of wild-type FHIT inhibited the expression of
endogenous cyclin D1 protein in HepG2 cells.
The results of the present study indicate that FHIT
is capable of directly inhibiting cell growth and suppressing
cyclin D1 expression in HepG2 cells. These observations provide a
rationale for targeting FHIT and the associated pathways as a novel
approach for chemoprevention and cancer therapy. Additional studies
are required to examine the clinical relevance of impairments in
the expression and function of FHIT with respect to HCC and other
types of human cancer.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81060204) to Lin Wang and
the Yunnan Natural Science Foundation (grant no. 2010CD080) to Lin
Wang.
References
|
1
|
Plymoth A, Viviani S and Hainaut P:
Control of hepatocellular carcinoma through hepatitis B vaccination
in areas of high endemicity: perspectives for global liver cancer
prevention. Cancer Lett. 286:15–21. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li H, Zhang Y, Su T, et al: Hint1 is a
haplo-insufficient tumor suppressor in mice. Oncogene. 25:713–721.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang L, Li H, Zhang Y, et al: HINT1
inhibits beta-catenin/TCF4, USF2 and NFkappaB activity in human
hepatoma cells. Int J Cancer. 124:1526–1534. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Negrini M, Monaco C, Vorechovsky I, et al:
The FHIT gene at 3p14.2 is abnormal in breast carcinomas. Cancer
Res. 56:3173–3179. 1996.
|
|
5
|
Bugert P, Wilhelm M and Kovacs G: FHIT
gene and the FRA3B region are not involved in the genetics of renal
cell carcinomas. Genes Chromosomes Cancer. 20:9–15. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sugimoto K, Yamada K, Miyagawa K, et al:
Decreased or altered expression of the FHIT gene in human
leukemias. Stem Cells. 15:223–228. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tanaka H, Shimada Y, Harada H, et al:
Methylation of the 5′ CpG island of the FHIT gene is closely
associated with transcriptional inactivation in esophageal squamous
cell carcinomas. Cancer Res. 58:3429–3434. 1998.
|
|
8
|
Croce CM, Sozzi G and Huebner K: Role of
FHIT in human cancer. J Clin Oncol. 17:1618–1624. 1999.PubMed/NCBI
|
|
9
|
Simon B, Bartsch D, Barth P, et al:
Frequent abnormalities of the putative tumor suppressor gene FHIT
at 3p14.2 in pancreatic carcinoma cell lines. Cancer Res.
58:1583–1587. 1998.PubMed/NCBI
|
|
10
|
Calvisi DF, Conner EA, Ladu S, et al:
Activation of the canonical Wnt/beta-catenin pathway confers growth
advantages in c-Myc/E2F1 transgenic mouse model of liver cancer. J
Hepatol. 42:842–849. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Behari J, Zeng G, Otruba W, et al:
R-Etodolac decreases beta-catenin levels along with survival and
proliferation of hepatoma cells. J Hepatol. 46:849–857. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Monga SP: Role of Wnt/beta-catenin
signaling in liver metabolism and cancer. Int J Biochem Cell Biol.
43:1021–1029. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee HS, Park MH, Yang SJ, et al: Novel
candidate targets of Wnt/beta-catenin signaling in hepatoma cells.
Life Sci. 80:690–698. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang L, Zhang Y, Li H, et al: Hint1
inhibits growth and activator protein-1 activity in human colon
cancer cells. Cancer Res. 67:4700–4708. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zöchbauer-Müller S, Fong KM, Maitra A, et
al: 5′ CpG island methylation of the FHIT gene is correlated with
loss of gene expression in lung and breast cancer. Cancer Res.
61:3581–3585. 2001.
|
|
16
|
Ohta M, Inoue H, Cotticelli MG, et al: The
FHIT gene, spanning the chromosome 3p14.2 fragile site and renal
carcinoma-associated t(3;8) breakpoint, is abnormal in digestive
tract cancers. Cell. 84:587–597. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zheng S, Ma X, Zhang L, et al:
Hypermethylation of the 5′ CpG island of the FHIT gene is
associated with hyperdiploid and translocation-negative subtypes of
pediatric leukemia. Cancer Res. 64:2000–2006. 2004.
|
|
18
|
Yang Q, Nakamura M, Nakamura Y, et al:
Two-hit inactivation of FHIT by loss of heterozygosity and
hypermethylation in breast cancer. Clin Cancer Res. 8:2890–2893.
2002.PubMed/NCBI
|
|
19
|
Leal MF, Lima EM, Silva PN, et al:
Promoter hypermethylation of CDH1, FHIT, MTAP and PLAGL1 in gastric
adenocarcinoma in individuals from Northern Brazil. World J
Gastroenterol. 13:2568–2574. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mirandola P, Gobbi G, Sponzilli I, et al:
TRAIL-induced apoptosis of FHIT-negative lung cancer cells is
inhibited by FHIT re-expression. J Cell Physiol. 220:492–498. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Werner NS, Siprashvili Z, Fong LY, et al:
Differential susceptibility of renal carcinoma cell lines to tumor
suppression by exogenous Fhit expression. Cancer Res. 60:2780–2785.
2000.PubMed/NCBI
|
|
22
|
Sevignani C, Calin GA, Cesari R, et al:
Restoration of fragile histidine triad (FHIT) expression induces
apoptosis and suppresses tumorigenicity in breast cancer cell
lines. Cancer Res. 63:1183–1187. 2003.PubMed/NCBI
|
|
23
|
Zheng H, Tsuneyama K, Takahashi H, et al:
Expression of PTEN and FHIT is involved in regulating the balance
between apoptosis and proliferation in lung carcinomas. Anticancer
Res. 27:575–581. 2007.PubMed/NCBI
|
|
24
|
Li H, Balajee AS, Su T, et al: The HINT1
tumor suppressor regulates both gamma-H2AX and ATM in response to
DNA damage. J Cell Biol. 183:253–265. 2008. View Article : Google Scholar : PubMed/NCBI
|