Introduction
The avian influenza virus is a member of the
Orthomyxoviridae family of the Influenzavirus A
genus. The subtypes capable of infecting humans are H5N1, H7N2,
H7N3, H7N7 and H9N2. The majority of avian influenza viruses lead
to mild symptoms in infected humans, which mainly include
conjunctivitis or upper respiratory tract infection, with the
exception of H5N1 infections, which have a mortality rate >50%
(1). A new reassortant viral
subtype named H7N9 was initially isolated from a patient with a
severe lower respiratory tract infection (2). Internal genes of this virus originate
from the H9N2 avian influenza virus (2). Patients who are infected with the
H7N9 virus usually present flu-like symptoms possibly accompanied
by a headache, muscle aches and general malaise. For patients with
severe infections, the progression of the disease is rapid. The
symptoms include severe pneumonia, hyperthermia (which is mostly
sustained at 39°C or above) and difficulty in breathing, and may
also be accompanied by hemoptysis. The symptoms may also progress
rapidly to acute respiratory distress syndrome and even lead to
mortality (3).
As of July 31, 2013, a total of 133 human cases
(including 44 cases of mortality) infected with the H7N9 virus had
been reported in 41 cities among 11 provinces in mainland China
(4). At present, cases occur
sporadically. It has been demonstrated that the H7N9 avian flu
virus can be transmitted to humans by avians, but with limited
human-to-human transmission activity (5). Due to the lack of vaccines and the
persistence of the H7N9 virus in chickens (6), there remains a risk of human
infection with the H7N9 virus. On April 27, 2013, the H7N9 virus
[A/Changsha/2/2013 (H7N9)] was detected from the tracheal aspirates
of a patient with severe pneumonia in Changsha, China. In the
present study, the whole genome of the A/Changsha/2/2013 virus was
sequenced, and the evolution and molecular characteristics of the
virus were analyzed.
Materials and methods
Patient
A 54-year-old male patient developed a fever and
sore throat on April 15, 2013. The diagnostic results indicated
that the patient was infected with the H7N9 avian influenza virus,
and infection with the H7N9 virus was subsequently confirmed. The
patient eventually succumbed to severe disease. This study was
approved by the ethics review board of Changsha Center for Disease
Control and Prevention (Changsha, China). Prior written and
informed consent were obtained from the patient’s family.
Reverse transcription (RT)-PCR
The A/Changsha/2/2013 (H7N9)-containing tracheal
aspirate samples were inactivated at 65°C for 30 min and then used
for the viral genome RNA extraction using an RNeasy®
Mini kit (Qiagen Inc., Hilden, Germany). Eight gene fragments
[polymerase basic protein 2 (PB2), PB1, polymerase acidic (PA),
hemagglutinin (HA), nucleoprotein (NP), neuraminidase (NA), matrix
protein (MP) and non-structural (NS)] of the new isolated H7N9
virus were amplified using the SuperScript® III One-Step
RT-PCR System with Platinum® Taq RT-PCR (Invitrogen Life
Technologies, Carlsbad, CA, USA). The reaction volume was 25 μl,
including 12.5 μl 2X reaction mix, 1.0 μl SuperScript™ III
RT/Platinum® Taq mix, 0.25 μl forward primer (20 μM), 0.25 μl
reverse primer (20 μM), 6.0 μl RNase-free water and 5.0 μl template
RNA sample. In total, 56 primers were used in the present study and
are shown in Table I. The PCR
products were sequenced by Takara Bio, Inc. (Dalian, China).
 | Table IPrimers used in the study. |
Table I
Primers used in the study.
| Primer | Sequence (5′-3′) | Product (bp) |
|---|
| HA-1-F |
ATGAACACTCAAATCCTGGT | 978 |
| HA-1-R |
TGCTAGCAGCAGACTCCTTT | |
| HA-2-F |
ACCAAGCTATATGGGAGTGG | 832 |
| HA-2-R |
CAAAGCAACCAGTGCCATCT | |
| HA-3-F |
TTGCCATTTCAGAACATAGA | 810 |
| HA-3-R |
GTAGAAACAAGGGTGTTTTT | |
| NA-1-F |
ATGAATCCAAATCAGAAGAT | 835 |
| NA-1-R |
CGTAACATGAGCATTCTTCA | |
| NA-2-F |
GTAGTATGGTACAACAGAAGG | 623 |
| NA-2-R |
TAGTCCATGAAAGATCCACT | |
| NA-3-F |
CAGATAGACCCAGTAGCAAT | 486 |
| NA-3-R |
CTCTATTTTAGCCCCATCAG | |
| NA-4-F |
ATGACCCTTATCCAGGTAAT | 426 |
| NA-4-R |
AGAATAAACAAGGGTCTTTT | |
| MP-1-F |
ATGAGTCTTCTAACCGAGGT | 767 |
| MP-1-R |
CTAGAGGCTCACTTGAACCG | |
| MP-2-F |
TACTACTAACCCACTAATTAGG | 502 |
| MP-2-R |
AGTAGAAACAAGGTAGTTTT | |
| NP-1-F |
ATGGCGTCTCAAGGCACCAA | 790 |
| NP-1-R |
GTGCAGACCTTGCCAGAAAA | |
| NP-2-F |
GCATATGAGAGAATGTGCAA | 818 |
| NP-2-R |
CCGAAGAAATAAGATCCTTCAT | |
| NP-3-F |
ACTCTGCAGCGTTTGAGGAC | 520 |
| NP-3-R |
AGTAGAAACAAGGGTATTTT | |
| NS-1-F |
ATGGATTCCAATACTGTGTC | 652 |
| NS-1-R |
ACTTTGTAGAGAGTGGAGATC | |
| NS-2-F |
TCAGTGTGATTTTCAATCGG | 464 |
| NS-2-R |
AGTAGAAACAAGGGTGTTTT | |
| NS-3-F |
TCAGTGTGATTTTCAATCGG | 355 |
| NS-3-R |
CAAAGCTATTCTCCGTAATT | |
| PA-1-F |
ATGGAAGACTTTGTGCGACA | 1000 |
| PA-1-R |
AATTGGGGTTTATGCCTTTC | |
| PA-2-F |
CTCCCTGCTCTCAGCGGTCGAAAT | 782 |
| PA-2-R |
TGGTTCCAACCTTGGGTCGG | |
| PA-3-F |
GCATCTTGTGCAGCCATGGA | 606 |
| PA-3-R |
CCTAAGCGCCTGAACAATGA | |
| PA-4-F |
CAATGGGACCTCCAAGATCA | 381 |
| PA-4-R |
AGGCACTCCTCGATTGCTTC | |
| PA-5-F |
CAATGGGACCTCCAAGATCA | 509 |
| PA-5-R |
AGTAGAAACAAGGTACTTTT | |
| PB1-1-F |
ATGGATGTCAATCCGACTTT | 962 |
| PB1-1-R |
ATTGCTAGAAACATCCGGGG | |
| PB1-2-F |
GCAAGCTGAAAAGGAGGGCA | 1000 |
| PB1-2-R |
TGCGTATCACCCCTGTGACA | |
| PB1-3-F |
TTCGTAGCTAACTTCAGTATG | 817 |
| PB1-3-R |
AGTAGAAACAAGGCATTTTT | |
| PB1-4-F |
TTCGTAGCTAACTTCAGTATG | 800 |
| PB1-4-R |
TTTTCATGAAGGACAAGCTA | |
| PB2-1-F |
ATGGAAAGAATAAAAGAACTAAG | 992 |
| PB2-1-R |
TTGAAAGTGAAACCTCCAAA | |
| PB2-2-F |
TAGAAGAGCAACAGTATCAGC | 1000 |
| PB2-2-R |
GAATAGAACCCTCACGAACC | |
| PB2-3-F |
CCATGATGTGGGAGATCAAT | 703 |
| PB2-3-R |
GGTCGTTTTTAAACAATTCG | |
| PB2-4-F |
CCATGATGTGGGAGATCAAT | 714 |
| PB2-4-R |
AGTAGAAACAAGGTCGTTTT | |
Phylogenetic analysis
The sequence data were edited and aligned using
BioEdit software (https://www.bioedit.com/). The full-genome sequences
of the virus isolated in the present study are available from
GenBank. The accession numbers of the sequences are: HA, KF420297;
NA, KF420299; MP, KF420301; NP, KF420303; NS, KF420305; PA,
KF420307; PB1, KF420309; and PB2, KF420311. Online blast analysis
demonstrated that all eight gene sequences of A/Changsha/2/2013
(H7N9) were highly homologous to those in the new H7N9 virus
strains isolated in mainland China and Taiwan in March-April 2013.
The homology was >99%, which indicated that the new isolated
virus was the H7N9 virus. The new H7N9 virus gene sequences
reported in the influenza virus database between March 1 and August
1, 2013, were retrieved for the construction of phylogenetic trees
(7). Phylogenetic trees were
constructed using the neighbor-joining method with MEGA software,
version 5.2 (http://www.megasoftware.net/megamac.php). The
molecular characteristics of each gene were analyzed.
Results
Phylogenetic analysis
The H7N9 virus nucleic acid test of the tracheal
aspirate samples from the male patient was positive and the virus
was identified as the H7N9 virus [A/Changsha/2/2013 (H7N9)]. The HA
gene phylogenetic tree (Fig. 1)
showed that the new H7N9 viruses isolated in 2013 were closely
genetically associated. All the viruses were in a large cluster,
with the exception of two Shanghai strains of the human H7N9 virus
(A/Shanghai/4655T/2013 and A/Shanghai/4665T/2013), which formed a
sub-branch. The A/Changsha/2/2013 virus in the present study was
closely associated with H7N9 viruses of different regions and
sources, including H7N9 viruses of human (A/Shanghai/4842T/2013),
avian (A/duck/Anhui/SC702/2013) and environmental
(A/environment/Nanjing/2913/2013) origin.
The NA gene phylogenetic tree (Fig. 2) was similar to that of the HA
gene. The sequences of the new H7N9 viruses were also in a large
cluster, while two Shanghai strains of the human H7N9 virus
(A/Shanghai/4664T/2013 and A/Shanghai/4821T/2013) formed a
sub-branch. The A/Changsha/2/2013 H7N9 virus was clustered together
with an environmental H7N9 virus
(A/environment/Shanghai/S1436/2013), pigeon H7N9 virus
(A/pigeon/Shanghai/S1423/2013) and human H7N9 virus
(A/Shanghai/4798T/2013) isolated in Shanghai, China.
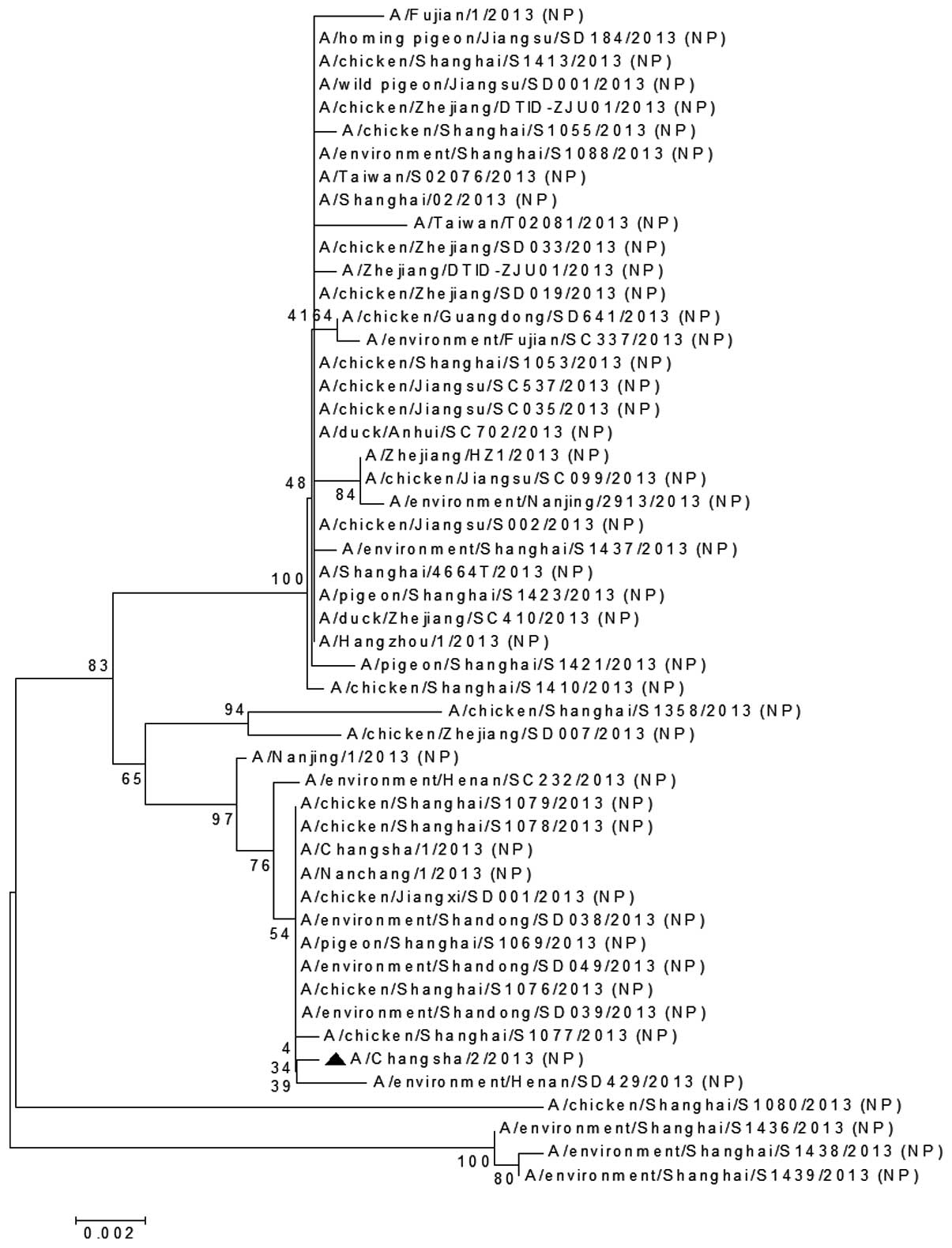
The NP (Fig. 3) and
NS genes (Fig. 4) of the H7N9
viruses isolated in 2013 formed a phylogenetic tree with two main
branches. One branch contained the avian, human and environmental
H7N9 viruses isolated from different regions. Another branch
contained Shanghai strains of environmental H7N9 viruses. The NP
gene of A/Changsha/2/2013 was clustered together on the phylogenic
tree with those from the chicken H7N9 virus isolated in Shanghai
(A/chicken/Shanghai/S1077/2013), the environmental H7N9 virus
strain isolated in Henan, China (A/environment/Henan/SD429/2013)
and other H7N9 viruses (Fig. 3).
The NS gene of the isolated H7N9 virus from the present case was
clustered together with the chicken H7N9 virus isolated in Shanghai
(A/chicken/Shanghai/S1076/2013) and the chicken H7N9 virus isolated
in Zhejiang, China (A/chicken/Zhejiang/SD033/2013; Fig. 4).
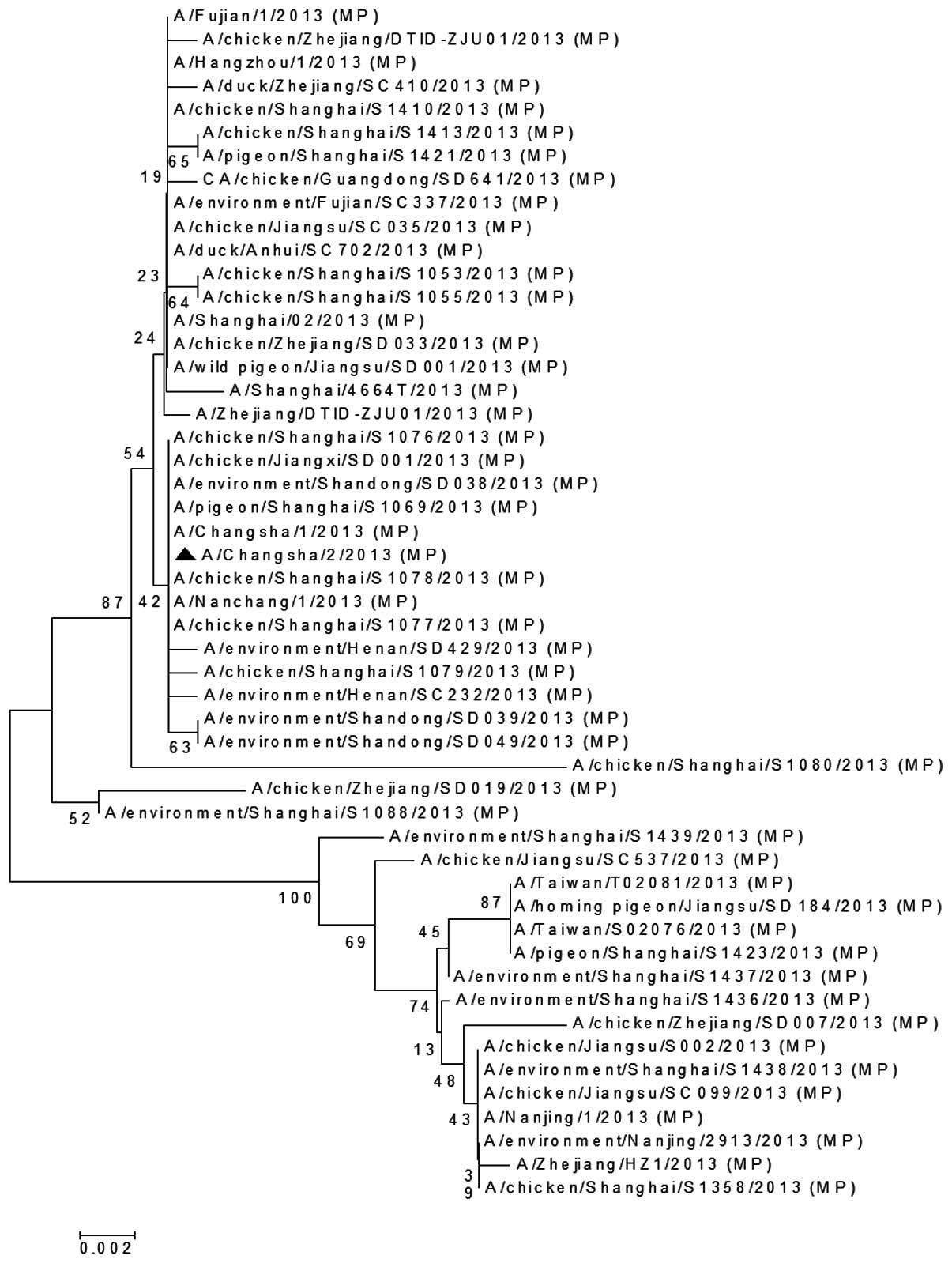
The MP (Fig. 5), PA
(Fig. 6), PB1 (Fig. 7) and PB2 (Fig. 8) genes of the H7N9 viruses isolated
in 2013 also formed phylogenetic trees with two main branches;
however, each branch contained avian, human and environmental H7N9
viruses of different regions. The phylogenetic tree demonstrated
that the MP gene of A/Changsha/2/2013 was closely associated with
the avian H7N9 virus isolated in Shanghai
(A/chicken/Shanghai/S1078/2013) and the human H7N9 virus isolated
in Nanchang, China (A/Nanchang/1/2013; Fig. 5). The PA gene of A/Changsha/2/2013
was clustered together on the phylogenic tree with the chicken H7N9
virus (A/chicken/Jiangsu/SC035/2013), chicken H7N9 virus
(A/chicken/Shanghai/S1077/2013) and human H7N9 virus
(A/Hangzhou/1/2013; Fig. 6). The
phylogenetic tree of the PB1 gene demonstrated that the
A/Changsha/2/2013 H7N9 virus was closely associated with certain
environmental H7N9 viruses (A/environment/Henan/SD429/2013 and
A/environment/Henan/SC232/2013; Fig.
7). The PB2 gene of A/Changsha/2/2013 was clustered together on
the phylogenic tree with environmental H7N9 viruses isolated in
Henan (A/environment/Henan/SD429/2013) and Shandong, China
(A/environment/Shandong/SD049/2013; Fig. 8). These results suggest that the
A/Changsha/2/2013 strain is evolutionally close to other H7N9 avian
influenza virus stains reported by Genbank between March and August
2013.
Characteristics of the viral genes
In order to analyze the key mutation sites, the
genome sequence of A/Changsha/2/2013 (H7N9) was compared with those
of the new H7N9 viruses reported in the influenza virus database in
2013. There was only one single amino acid arginine (R) at the
protein cleavage site (amino acids 339–345) of the linker peptide
between HA1 and HA2 of the A/Changsha/1/2013 (H7N9) HA gene, which
was similar to those in other newly isolated H7N9 viruses,
indicating that the virus had a low pathogenicity for poultry
(5). The Q226L amino acid mutation
(the amino acid site encoded by the H3 type influenza virus
corresponded to amino acid 235 in the present study) occurred in
the HA protein of the new H7N9 viruses of human, avian and
environmental origin. The mutation rate was 88.85% (46/52). G228S
and G186V mutations were also revealed to be present at the
receptor binding site, with a mutation rate of 100%. However, no
A138S mutation was identified (Table
II). The PB2 gene of eight viruses among 51 new H7N9 viruses
harbored the E627K mutation. However, only one virus harbored the
D701N mutation. No E627K and D701N mutations were detected in the
A/Changsha/2/2013 PB2 protein (Table
II). Amino acid deletions were also identified in the NA coding
protein at amino acids 69–73, and the NS1-coding protein at amino
acids 218–230 of all the new H7N9 viruses that were isolated in
2013, respectively. Resistance gene loci analysis showed that the
R294K mutation did not occur in the NA coding protein of the
A/Changsha/2/2013 virus and other 50 H7N9 virus strains. However,
it occurred in a human H7N9 virus strain isolated in Taiwan. The
S31N mutation of the M2 protein encoded by the MP gene was
identified in the 52 new H7N9 virus strains. Other mutations in the
new H7N9 virus isolated in 2013 included: P42S in the NS1 protein
encoded by the NS gene; N30D and T215A in the M1 protein encoded by
MP gene; and L89V in the PB2 protein (Table II). These results suggest that the
A/Changsha/2/2013 H7N9 virus strain has molecular characteristics
that may facilitate adaptation of the virus to mammalian hosts and
may even bind to human receptors.
| Table IIMolecular analysis of important amino
acids in the HA, NA, NS1, M1, M2, PB1 and PB2 proteins of the H7N9
virus. |
Table II
Molecular analysis of important amino
acids in the HA, NA, NS1, M1, M2, PB1 and PB2 proteins of the H7N9
virus.
| Gene | Site | Position |
A/Changsha/2/2013 | Mutated virus | Non-mutated
virus |
|---|
| HA | Q226L | 226/235a | L | 46/52 | 6/52 |
| G228S | 228/237a | G | 52/52 | 0/52 |
| A138S | 138/130a | A | 0/52 | 52/52 |
| G186V | 186/195a | V | 52/52 | 0/52 |
| NA | R294K | 294/289b | R | 1/52 | 51/52 |
| | 69–73 | Deletion | 52/52 deletion | 0/52 no
deletion |
| NS1 | P42S | 42 | S | 50/50 | 0/50 |
| | 218–230 | Deletion | 50/50 deletion | 0/50 no
deletion |
| M1 | N30D | 30 | D | 50/50 | 0/50 |
| T215A | 215 | A | 50/50 | 0/50 |
| M2 | S31N | 31 | N | 50/50 | 0/50 |
| PB1 | H99Y | 99 | H | 0/48 | 48/48 |
| PB2 | L89V | 89 | V | 51/51 | 0/51 |
| E627K | 627 | E | 8/51 | 43/51 |
| D701N | 701 | D | 1/51 | 50/51 |
Discussion
The phylogenetic tree results demonstrated that the
HA and NA genes of the A/Changsha/2/2013 (H7N9) virus were closely
associated with those of the other new H7N9 viruses isolated in
2013. All the viruses were in a large cluster, while the two
Shanghai strains of the human H7N9 virus (A/Shanghai/4664T/2013 and
A/Shanghai/4821T/2013) formed a sub-branch. The results indicate
that the new human H7N9 viruses were highly homologous to avian and
environmental H7N9 viruses. It further confirmed that the H7N9
virus was transformed from poultry to humans. More H7N9 viruses
were isolated in different birds and environments, suggesting that
H7N9 viruses have been distributed in these avian species and
environments. The NP and NS genes of the H7N9 viruses isolated in
2013 formed a phylogenetic tree with two main branches. One branch
contained avian, human and environmental H7N9 viruses isolated in
different regions. Another branch contained three environmental
H7N9 viruses isolated in Shanghai. The MP, PA, PB1 and PB2 genes of
the H7N9 viruses formed phylogenetic trees with two main branches
and each branch contained avian, human and environmental H7N9
viruses isolated in different regions. This suggests that NP, NS,
MP, PA, PB1 and PB2 genes of the new H7N9 viruses isolated in 2013
are in different degrees of evolutionary separation. The different
branches indicate the genetic diversity of the H9N2 viruses from
which the genes originate (8).
The HAs of human influenza viruses bind to cell
sialic acid linked to galactose by an α-2,6 linkage, as found on
human cells, while avian viruses have a predilection for sialic
acid linked to galactose by α-2,3 linkages, as found on avian
epithelia. This receptor specificity is considered to be one of the
factors responsible for the species barrier that prevents avian
viruses from readily infecting humans. Avian influenza viruses
typically contain Gln226 in the HA gene encoding protein, while
human viruses contain Leu226 (8–11).
The HA protein of A/Changsha/2/2013 and other 45 other new human,
avian and environmental H7N9 viruses harbored the Q226L mutation
(with a mutation rate of 88.5%) in the receptor binding site. In
addition, 52 H7N9 viruses harbored the G186V and G228S mutations in
the receptor binding site of the HA protein. This indicates that
A/Changsha/2/2013 and other new H7N9 viruses specifically bind to
human receptors and are able to infect humans (8–10,12).
The E627K mutation of the PB2 polymerase protein was
identified in H5N1 and H7N7 avian influenza viruses isolated from
several cases of mortality. The E627K and D701N mutations favor the
efficient replication and dissemination of avian influenza viruses
in mammals (2,13–15).
Although no E627K and D701N mutations were detected in the PB2
protein of A/Changsha/2/2013, the E627K mutation and D701N mutation
occurred in eight human H7N9 virus strains and one human H7N9 virus
strain among the 51 new H7N9 viruses, respectively. However avian
and environmental H7N9 viruses had no E627K and D701N mutations.
This demonstrates that the new H7N9 viruses isolated in 2013
enhanced the adaptive capacity in the mammalian host. However,
further investigation is required.
A deletion at amino acids 69–73 in the NA protein
has been reported to increase viral virulence in mammals (11), while a deletion at amino acids
218–230 in the NS1 protein has been reported to reduce viral
virulence in mammals (16). The NA
and NS1 proteins of all the new H7N9 viruses isolated in 2013,
including A/Changsha/1/2013, have deletions at amino acids 69–73
and 218–230, respectively. Resistance gene loci analysis
demonstrated that the R294K mutations of the NA coding protein only
occurred in one H7N9 virus strain isolated in Taiwan
(A/Taiwan/S02076/2013), suggesting that A/Changsha/2/2013 and 50
other new H7N9 viruses isolated in 2013 were sensitive to
neuraminidase inhibitors (e.g. oseltamivir) (17). As neuraminidase inhibitors are
widely used in the treatment of H7N9 virus infection, the possible
development of drug resistance requires close monitoring. All 52
H7N9 viruses were resistant to the M2 ion channel blocker
amantadine. A/Changsha/2/2013 (H7N9) was isolated from a severe,
fatal case of pneumonia.
The main genetic characteristic of the
A/Changsha/2/2013 (H7N9) virus is that it is able to infect humans.
The new H7N9 viruses are distributed in different avian species and
environments and are capable of infecting humans. This suggests
that the next phase of H7N9 avian influenza prevention and control
is urgently required.
Acknowledgements
This study was supported by the Medical and Health
Research Project of Hunan, China (grant no. B2013-126).
References
|
1
|
To KK, Ng KH, Que TL, et al: Avian
influenza A H5N1 virus: a continuous threat to humans. Emerg
Microbes Infect. 1:e252012. View Article : Google Scholar
|
|
2
|
Gao R, Cao B, Hu Y, et al: Human infection
with a novel avian-origin influenza A (H7N9) Virus. N Engl J Med.
368:1888–1897. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wu S, Wu F, He J, et al: Emerging risk of
H7N9 influenza in China. Lancet. 381:1539–1540. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
H7N9 bird flu infections in humans in
China. http://www.moh.gov.cn/jkj/s7915v/201308/b4961105d78f4726a18dc3f8e53ac78e.shtmluri.
Accessed August 9, 2013
|
|
5
|
Qi X, Qian YH, Bao CJ, et al: Probable
person to person transmission of novel avian influenza A (H7N9)
virus in Eastern China, 2013: epidemiological investigation. BMJ.
347:f47522013. View Article : Google Scholar
|
|
6
|
Chen Y, Liang W, Yang S, et al: Human
infections with the emerging avian influenza A H7N9 virus from wet
market poultry: clinical analysis and characterisation of viral
genome. Lancet. 381:1916–1925. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Influenza Virus Database. http://www.ncbi.nlm.nih.gov/genomes/FLU/Database/nph-select.cgi?uriAccessed
August 4, 2013
|
|
8
|
Liu D, Shi W, Shi Y, et al: Origin and
diversity of novel avian influenza A H7N9 viruses causing human
infection: phylogenetic, structural, and coalescent analyses.
Lancet. 381:1926–1932. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Srinivasan K, Raman R, Jayaraman A,
Viswanathan K and Sasisekharan R: Quantitative description of
glycan-receptor binding of influenza A virus H7 hemagglutinin. PLoS
One. 8:e495972013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nidom CA, Takano R, Yamada S, et al:
Influenza A (H5N1) viruses from pigs, Indonesia. Emerg Infect Dis.
16:1515–1523. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Matsuoka Y, Swayne DE, Thomas C, et al:
Neuraminidase stalk length and additional glycosylation of the
hemagglutinin influence the virulence of influenza H5N1 viruses for
mice. J Virol. 83:4704–4708. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang H, Chen LM, Carney PJ, et al:
Structures of receptor complexes of a North American H7N2 influenza
hemagglutinin with a loop deletion in the receptor binding site.
PLoS Pathog. 6:e10010812010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Munster VJ, de Wit E, van Riel D, et al:
The molecular basis of the pathogenicity of the Dutch highly
pathogenic human influenza A H7N7 viruses. J Infect Dis.
196:258–265. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hatta M, Gao P, Halfmann P, et al:
Molecular basis for high virulence of Hong Kong H5N1 influenza A
viruses. Science. 293:1840–1842. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kageyama T, Fujisaki S, Takashita E, et
al: Genetic analysis of novel avian A(H7N9) influenza viruses
isolated from patients in China, February to April 2013. Euro
Surveill. 18:204532013.PubMed/NCBI
|
|
16
|
Jackson D, Hossain MJ, Hickman D, Perez DR
and Lamb RA: A new influenza virus virulence determinant: the NS1
protein four C-terminal residues modulate pathogenicity. Proc Natl
Acad Sci USA. 105:4381–4386. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kiso M, Ozawa M, Le MT, et al: Effect of
an asparagine-to-serine mutation at position 294 in neuraminidase
on the pathogenicity of highly pathogenic H5N1 influenza A virus. J
Virol. 85:4667–4672. 2011. View Article : Google Scholar : PubMed/NCBI
|