Introduction
Gene therapy has been defined as the deliberate
introduction of genetic material into cells in order to treat or
prevent a disease. The success of gene therapy ultimately depends
on gene delivery vectors. Thus, a number of studies have focused on
creating more efficient, stable and safe gene therapy vectors.
Viral and non-viral vectors are the two main groups of vectors that
are used in gene therapy. To date, ~70% of clinical trials and the
majority of studies have used viral vectors. Widely used viral
vectors include adenovirus, adeno-associated virus, retrovirus and
lentivirus. However, these vectors possess limitations, including
possible safety problems (1,2) and
challenges for large scale production. In contrast to the commonly
employed viral vectors, baculovirus has unique properties of
inherent safety, ease and speed of virus generation in high
quantities, low cytotoxicity and extreme transgene capacity
(3). Thus, the baculovirus vector
is being increasingly applied and continually developed for
efficient gene therapy.
The baculovirus expression vector system (BEVS) is
recognized as a feasible and safe technology to produce recombinant
proteins in insect cells, which are eukaryotic. The basis of BEVS
lies in large enveloped DNA viruses derived from insects. The
prototype virus is Autographa californica multiple
nucleopolyhedrovirus (AcMNPV), which uses a 134-kbp genome to
encode 156 viral proteins. AcMNPV is occluded in a polyhedrin (PH)
and p10 proteins and expression of the virus is under the control
of PH and p10 promoters, respectively. However, as these promoters
are not indispensable for viral replication, the PH or p10 promoter
may be replaced by other promoters. Infection of insect cells with
a virus encoding a desired transgene under the control of the
powerful baculovirus PH or p10 promoter leads to recombinant
protein production in high quantities (4). In 1995, it was demonstrated that a
recombinant baculovirus was able to infect mammalian cells and
express foreign genes under the control of a mammalian promoter
(5). Since then, numerous cells
have been shown to be permissive for baculovirus-mediated gene
delivery, including animal cells from humans, cows, rodents, pigs,
rabbits, fish or with an avian origin, as well as a number of
primary cells, including embryonic, adult and induced pluripotent
stem cells (6).
The Bac-to-Bac Baculovirus Expression system
(Invitrogen Life Technologies, Carlsbad, CA, USA) provides a rapid
and efficient method for generating recombinant baculoviruses
(7). A number of pFastBac™
vectors, including pFastBac 1, HT and Dual, may be used in this
system. The pFastBac 1 and HT vectors have a strong PH promoter for
high-level protein expression, while the pFastBac Dual vector is a
non-fusion vector that has two strong baculovirus promoters, PH and
p10, which allow the simultaneous expression of two proteins.
Previously, PH or p10 promoters have been replaced by mammalian
promoters in one of the aforementioned pFastBac vectors. However,
to date, to the best of our knowledge, there have been no studies
in which the PH and p10 promoters in the pFastBac Dual vector have
been replaced with mammalian promoters. Originally, the pFastBac
Dual vector allowed the production of two proteins in insect cells
only. The aim of the present study was to investigate the effect of
replacing the two promoters with two strong mammalian promoters and
to determine whether the vector was effective in coexpressing two
target proteins in mammalian cells. If successful, this would
expand the application of the vector from protein expression in
insect cells to dual-gene delivery in vitro and in
vivo.
Coexpression of two or more exogenous genes in an
organism becomes increasingly significant with genetic engineering
since the metabolic pathways and physiological processes are
complex and require the coordination functions of a variety of
genes. Currently, widely used multiexpression strategies include
coinfection (8), internal ribosome
entry sites (IRES) (9), fusion
protein (10) and a self-cleaving
2A peptide (11). However, these
strategies have disadvantages. Firstly, the correlation between the
expression levels of the two genes is usually hard to examine in
the coinfection approach. Secondly, IRES elements can be large and
attenuate the expression of downstream genes. Thirdly, fusion
protein production may result in a compromised function, which may
potentially be due to improper protein folding or trafficking.
Finally, cloning vectors harboring a 2A peptide gene are not
publicly available. One method to overcome these problems is the
use of a dual-expression vector that includes two promoters. In
addition, multi-gene therapy is an inevitable trend in gene therapy
development as it provides a more rational and superior approach
compared with single-gene therapy. Due to the advantages of the
Bac-to-Bac Baculovirus Expression system, the pFastBac Dual vector
was selected for the current study. However, this vector can only
express proteins in insect cells. Thus, in the present study, a
pFastBac Dual vector was developed that possessed a gene cassette
consisting of the enhanced green fluorescent protein (EGFP) gene
and glial cell line-derived neurotrophic factor (GDNF) gene. The
two genes were under the control of the cytomegalovirus (CMV)
promoter and generated a recombinant baculovirus named Bac
Dual-CMV-EGFP-CMV-GDNF. Human embryonic kidney (HEK) 293T cells and
HeLa cells were transduced with this recombinant baculovirus.
Indirect immunofluorescence was applied to demonstrate EGFP and
GDNF expression in a single cell simultaneously and western blot
analysis was performed to detect the expression of the protein of
interest. Therefore, the present study focused on developing a
dual-promoter and coexpressing vector.
Materials and methods
Main reagents
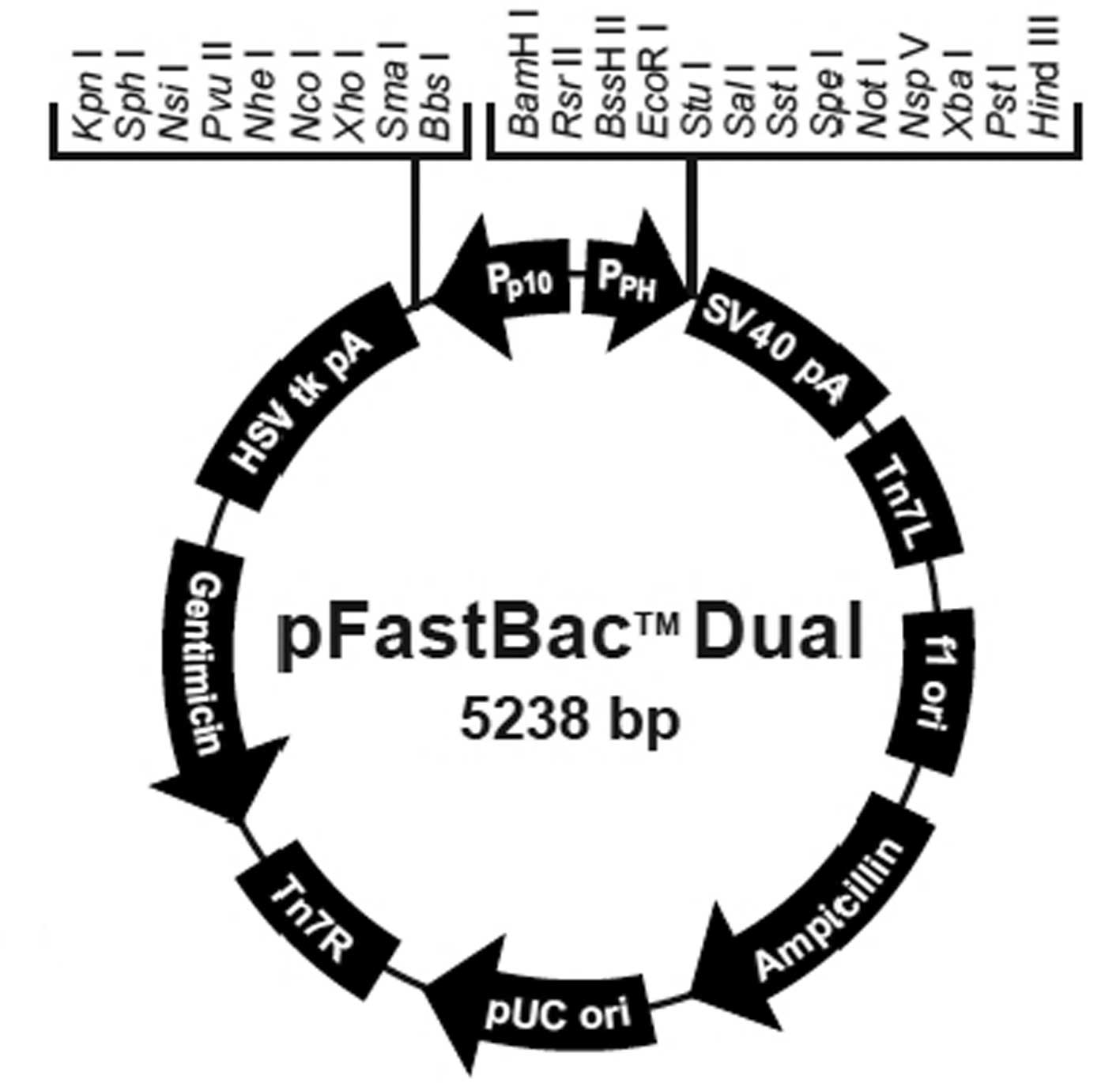
The pFastBac Dual vector (Fig. 1), DH10Bac Escherichia coli (E.
coli) cells, Sf9 cells, PureLink HiPure Plasmid Midiprep kit,
Lipofectamine 2000, Cellfectin II reagent, fetal bovine serum
(FBS), Dulbecco’s modified Eagle’s medium (DMEM) and Sf-900 III
serum-free medium were purchased from Invitrogen Life Technologies
(Shanghai, China). DH5α E. coli competent cells, restriction
endonuclease, T4 DNA ligase, LA Taq with GC buffer and DNA
markers were products of Takara Bio, Inc. (Dalian, China). DNA
extraction and plasmid DNA purification kits were obtained from
Qiagen (Germantown, MD, USA). Rat GDNF and pEGFP-1 plasmids were
conserved in the laboratory at Rui Jin Hospital (Shanghai, China).
Anti-GDNF rabbit polyclonal antibodies were purchased from Santa
Cruz Biotechnologies, Inc. (Santa Cruz, CA, USA). DyLight594 goat
anti-rabbit IgG was purchased from MultiSciences Biotech Co., Ltd.
(Hangzhou, China). Horseradish peroxidase was purchased from
Beyotime Institute of Biotechnology (Shanghai, China). Primers were
synthesized and DNA was sequenced by Invitrogen Life Technologies
(Shanghai, China).
Cell lines and culture
Sf9 cells were propagated at 27°C in Sf-900 III
serum-free medium. HeLa and HEK 293T cells, provided by the Cell
Bank of the Chinese Academy of Science (Shanghai, China), were
cultured in DMEM supplemented with 10% FBS in a humidified
environment with 5% CO2 at 37°C.
Construction of the pFastBac
Dual-CMV-EGFP-CMV-GDNF plasmid
Polymerase chain reaction (PCR) primers, F1 and R1,
were designed for CMV-EGFP (Table
I). The BamHI gene (GGATCC) was added to the 5′ end of
primer F1, while the KpnI gene (GGTACC) and terminator codon
(TTA) were added to the 5′ end of the R1 primer. Next, a CMV-EGFP
fragment was amplified from the pEGFP-C1 vector by PCR with the F1
and R1 primers. The CMV-EGFP fragment and pFastBac Dual vector were
digested simultaneously with BamHI and KpnI and then
connected with T4 DNA ligase following electrophoresis and
recovery. E. coli DH5α cells were transformed with 5 μl
connected products. A single colony was selected and the pFastBac
Dual-CMV-EGFP plasmid was obtained following verification by gene
sequencing.
 | Table IPrimers used for the amplification of
CMV-EGFP and CMV-GDNF and the analysis of recombinant bacmid by
PCR. |
Table I
Primers used for the amplification of
CMV-EGFP and CMV-GDNF and the analysis of recombinant bacmid by
PCR.
| Primer name | Sequence, 5′→3′ |
|---|
| F1 |
GCCGCCGGATCCTAGTTATTAATAGTAATCAATTACGGGGTCA |
| R1 |
ACCACCGGTACCTTACTTGTACAGCTCGTCCATGCC |
| F2 |
GCCGCCGTCGACTAGTTATTAATAGTAATCAATTACGGGGTCA |
| R2 |
CGACATCCCATAACTTCATGGTGGCGATCTGACGGTTCACTAAACCAGCT |
| F3 |
TTTAGTGAACCGTCAGATCGCCACCATGAAGTTATGGGATGTCGTGGCT |
| R3 |
ACCACCAAGCTTTCAGATACATCCACACCGTTTAGC |
| pUC/M13 forward |
CCCAGTCACGACGTTGTAAAACG |
| pUC/M13 reverse |
AGCGGATAACAATTTCACACAGG |
| Specific primer |
ATGAAGTTATGGGATGTCGTGG |
PCR splicing primers, F2, R2, F3 and R3, were
designed for CMV-GDNF (Table I).
The SalI gene (GTCGAC) was added to the 5′ end of primer F2
and the HindIII gene (AAGCTT) was added to the 5′ end of
primer R3. The CMV fragment was amplified from the pEGFP-C1 vector
by PCR with the F2 and R2 primers, while the GDNF fragment was
amplified from the GDNF plasmid with the F3 and R3 primers. Next,
the CMV and GDNF fragments were connected by overlap extension PCR
with the F2 and R3 primers and the CMV-GDNF fragment was obtained
following the recovery of the PCR products.
The pFastBac Dual-CMV-EGFP vector and CMV-GDNF
fragment were digested simultaneously with SalI and
HindIII and connected with T4 DNA ligase following
electrophoresis and recovery. Next, 5 μl connected products was
transformed into E. coli DH5α cells. A single colony was
selected and the pFastBac Dual-CMV-EGFP-CMV-GDNF plasmid was
obtained following verification by 1.2% agarose gel electrophoresis
and sequencing.
Generation of recombinant
baculovirus
Following the construction of the pFastBac
Dual-CMV-EGFP-CMV-GDNF plasmid, it was transformed into DH10Bac
E. coli cells for transposition into a bacmid. A white
colony and a blue colony were selected by blue/white colony
selection and the PureLink HiPure Plasmid Midiprep kit was used to
purify recombinant bacmid DNA. To verify the presence of the gene
of interest in the recombinant bacmid from the white colony, the
specific, pUC/M13 forward and reverse primers were synthesized
(Table I). PCR was then performed
with pUC/M13 primers or with the pUC/M13 forward and specific
primers. Correspondingly, PCR was performed with pUC/M13 primers
and the bacmid from the blue colony. The obtained reaction products
were analyzed by agarose gel electrophoresis.
Transfection of Sf9 cells with the recombinant
bacmid and Cellfectin II was performed according to the
manufacturer’s instructions. Cell morphology was observed daily to
view the characteristics of viral infection following transfection.
After 7 days, the supernatant was collected and the P1 viral stock
was obtained. The P2 viral stock was obtained following the
amplification of the P1 viral stock. To determine the titer of the
baculoviral stock, a viral plaque assay was performed.
Transfection of HEK 293T cells with
Lipofectamine 2000
One day prior to transfection, HEK 293T cells were
plated in a 24-well plate coated with poly-L-lysine in DMEM without
antibiotics with the result that the cells were 80–90% confluent at
the time of transfection. Next, the recombinant plasmid was
transfected into HEK 293T cells with Lipofectamine 2000, according
to the manufacturer’s instructions, to prepare for
immunofluorescence.
Transduction of HeLa cells with
recombinant baculovirus
HeLa cells were seeded in a 24- or 6-well plate with
DMEM and 10% FBS for 24 h. The medium was replaced with
phosphate-buffered saline (PBS) just prior to virus transduction.
Recombinant baculovirus was then added at a multiplicity of
infection of 200. Cells treated with PBS only were used as a
negative control. Cells were incubated at 37°C for 4 h following
treatment and the medium, including the virus, was replaced with
DMEM and 10% FBS. After 24 h, the cells in the 24- or 6-well plates
were analyzed by immunofluorescence and western blotting.
Immunofluorescence test
Following the transfection of HEK 293T cells with
Lipofectamine 2000 and the transduction of HeLa cells in a 24-well
plate with recombinant baculovirus, an indirect immunofluorescence
test was performed with a 1:300 dilution of anti-GDNF rabbit
polyclonal antibodies and a 1:300 dilution of DyLight594 goat
anti-rabbit IgG. The cells were observed with a fluorescence
microscope (Olympus Corporation, Tokyo, Japan).
Western blot analysis
Following the transduction of HeLa cells in a 6-well
plate with recombinant baculovirus, the cells were collected and
protein extracts were prepared in a lysis buffer. Western blot
analysis was then performed by incubating the filtrate with a 1:500
dilution of anti-GDNF rabbit polyclonal antibodies in Tris-buffered
saline Tween 20 at 4°C overnight. Next, a 1:1,000 dilution of
secondary antibodies conjugated to horseradish peroxidase was added
for 1 h at room temperature. Protein bands were treated using an
enhanced chemiluminescence assay kit (PerkinElmer, Inc., Waltham,
MA, USA).
Results
Verification of the recombinant
plasmid
Following the construction of the pFastBac
Dual-CMV-EGFP-CMV-GDNF plasmid, the size was evaluated by 1.2%
agarose gel electrophoresis. As shown in Fig. 2, the electrophoresis stripe in lane
1 was at the correct site (~7,426 bp). In addition, the sequencing
results demonstrated that the inserted gene fragment was in
accordance with expectations and there was no mutation (data not
shown). Therefore, the pFastBac Dual-CMV-EGFP-CMV-GDNF plasmid was
constructed successfully.
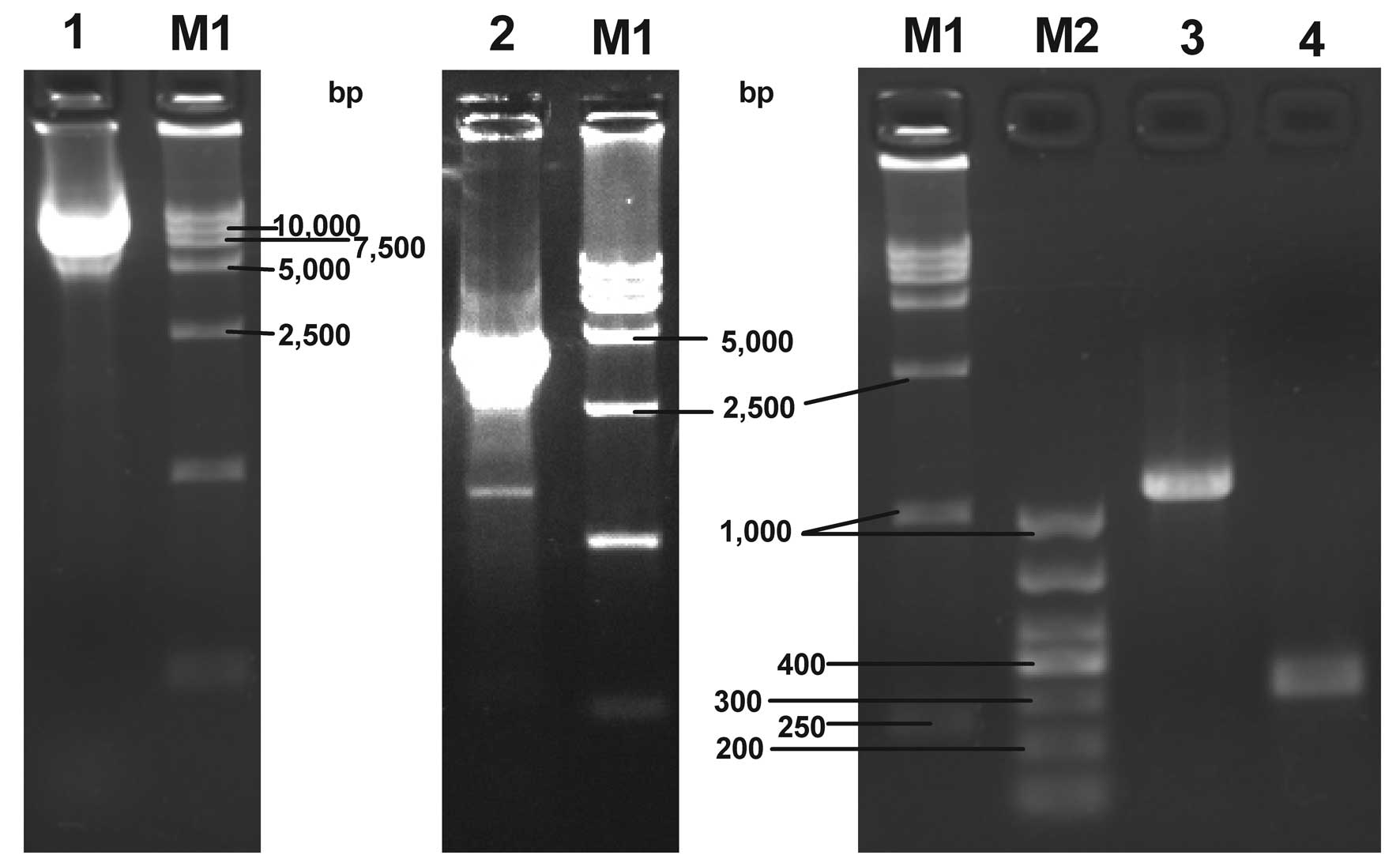 | Figure 2Agarose gel electrophoresis of the
recombinant plasmid and bacmid DNA PCR products. Lane 1, pFastBac
Dual-CMV-EGFP-CMV-GDNF plasmid; lane 2, PCR product of recombinant
bacmid DNA amplified with pUC/M13 forward and reverse primers; lane
3, PCR product of recombinant bacmid DNA amplified with the
specific and pUC/M13 reverse primers; lane 4, PCR product of
non-recombinant bacmid DNA amplified with pUC/M13 forward and
reverse primers; lane M1, DL 15,000 DNA marker; lane M2, DL 1,000
DNA marker; PCR, polymerase chain reaction; CMV, cytomegalovirus;
EGFP, enhanced green fluorescent protein; GDNF, glial cell
line-derived neurotrophic factor. |
Generation and confirmation of the
recombinant bacmid
As shown in lanes 2 and 3 of Fig. 2, PCR products of the expected sizes
were obtained (~4,800 bp and 1,200 bp) from the white colony,
indicating the presence of the gene of interest in the recombinant
bacmid. pUC/M13 forward and reverse primers were selected to
amplify the bacmid DNA from the blue colony and a PCR product of
the expected size (~300 bp) was achieved, as shown in lane 4
(Fig. 2). This demonstrated that
no transposition occurred in the blue colony. Thus, the results
indicated that transposition occurred in the white colony and the
expected recombinant bacmid was isolated.
Generation of the recombinant
baculovirus
Following transfection with Cellfectin II, Sf9 cells
typically exhibited characteristics of infected cells emerging in
succession as follows: Increased cell diameter, increased nuclei
size, cessation of cell growth, granular appearance, detachment and
cell lysis. These qualities demonstrated that Bac
Dual-CMV-EGFP-CMV-GDNF was constructed successfully. In addition,
the viral plaque assay revealed that the titer of the baculoviral
stock was 8×107 pfu/ml.
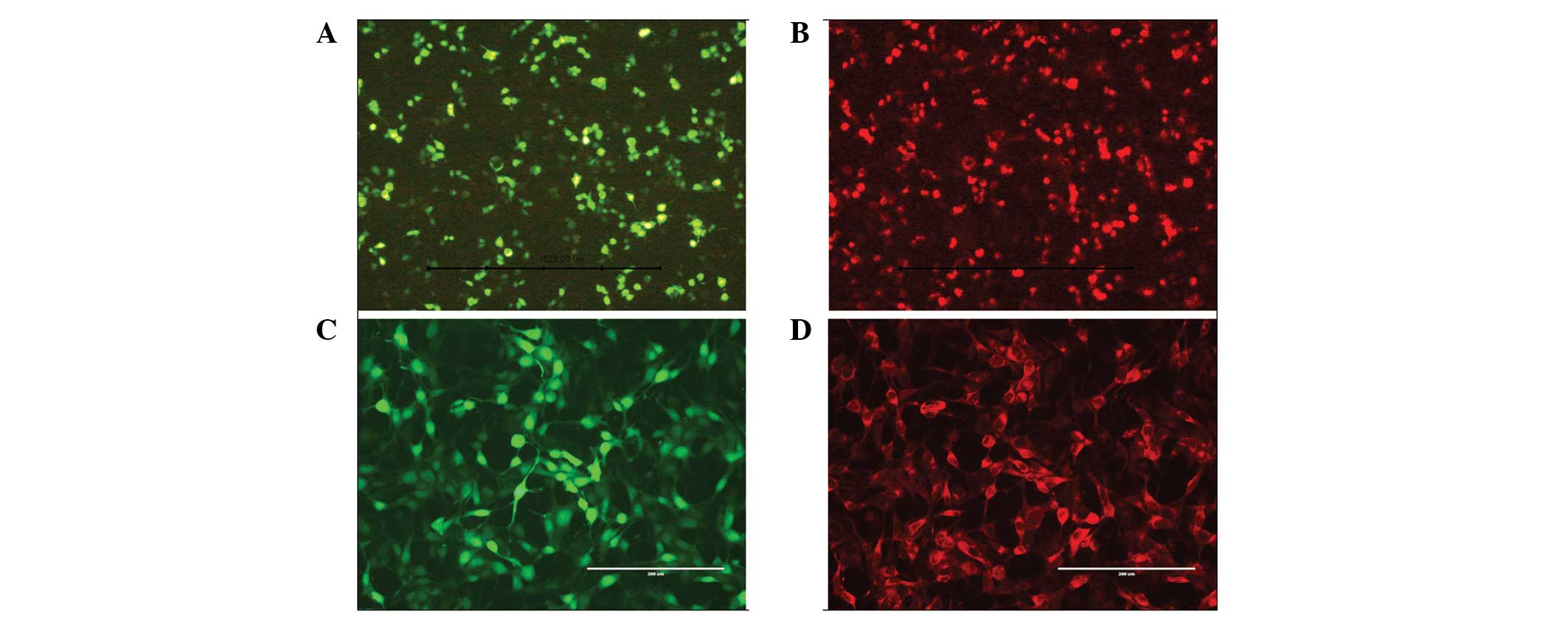
Identification by immunofluorescence
Immunofluorescence analysis indicated that EGFP and
GDNF were simultaneously expressed in the same HEK 293T cell
transfected with recombinant plasmid and Lipofectamine 2000
(Fig. 3A and B), confirming the
accuracy and usability of the recombinant plasmid. The synchronous
expression of EGFP and GDNF in a single HeLa cell transduced by
recombinant baculovirus demonstrated that the pFastBac Dual vector,
under the control of two mammalian promoters, was able to coexpress
two target genes in mammalian cells (Fig. 3C and D).
Western blot analysis
As shown in Fig 4,
western blot analysis results revealed no protein band in lane 2.
However, a distinct protein band with a relative molecular weight
of ~15 kDa corresponding to GDNF was observed in lane 1 (Fig. 4). This confirmed that GDNF was not
expressed in the non-transduced HeLa cells, while GDNF was
expressed in the HeLa cells transduced by recombinant baculovirus.
Therefore, western blot analysis demonstrated that GDNF was
expressed accurately in cells transduced with
Bac-CMV-EGFP-CMV-GDNF.
Discussion
GDNF was first identified as a potent survival
factor for midbrain dopaminergic neurons, but was then shown to be
a potent neurotrophic factor that had restorative effects in a wide
variety of rodent and primate models of Parkinson’s disease
(12). GDNF is broadly expressed
and essential for the development of the kidney and the enteric
nervous system. As a multifunctional protein, GDNF has the ability
to induce cellular survival, proliferation, migration and
differentiation (13). To date,
the scope of GDNF applications has been greatly expanded and
includes the treatment of Huntington’s disease, amyotrophic lateral
sclerosis, chronic pain, depression and addiction, as well as the
regeneration of the sciatic nerve (14). In view of its numerous
applications, GDNF was selected as the target protein in the
present study.
In this study, the PH and p10 promoters in the
pFastBac Dual vector were replaced by CMV promoters, whose
directions were consistent with the initial direction of the PH or
p10 promoter. A CMV-EGFP gene fragment was inserted into the
initial region of the p10 promoter gene and a CMV-GDNF gene
fragment, connected by the CMV promoter and GDNF genes, was cloned
into the initial site of the PH promoter gene. Following the
successful construction of the pFastBac Dual-CMV-EGFP-CMV-GDNF
plasmid, it was transfected into HEK 293T cells with Lipofectamine
2000. Through indirect fluoroimmunoassay technology, EGFP and GDNF
were shown to be expressed in a single HEK 293T cell at the same
time, which confirmed the accuracy and usability of the recombinant
plasmid. Next, Bac Dual-CMV-EGFP-CMV-GDNF was generated using the
Bac-to-Bac Baculovirus Expression system. A viral plaque assay
demonstrated that the virus titer was 8×107 pfu/ml. HeLa
cells were transduced with the recombinant baculovirus. Similarly,
the simultaneous existence of EGFP and GDNF in the same HeLa cell
was demonstrated by fluorescence detection and GDNF protein was
identified by western blot analysis. Therefore, in the present
study, a dual-promoter recombinant baculovirus vector was
constructed and shown to coexpress EGFP and GDNF in mammalian
cells.
Considering that GDNF must exist when EGFP is
expressed in the gene expression of Bac-CMV-EGFP-CMV-GDNF, the
position and quantity of expressed GDNF can be conveniently
estimated through observing the expression of EGFP. If the EGFP
gene were to be replaced with an imaging reporter gene,
non-invasive imaging of the reporter gene products would reveal the
temporal and spatial biodistribution of GDNF or other genes of
interest in virtually any location within living subjects.
Molecular imaging of reporters for target genes plays a vital role
in optimizing gene therapy by quantitatively imaging reporter gene
expression and the therapeutic effect of transgenes in vivo.
The sodium iodide symporter (NIS) has emerged as one of the most
promising reporter genes in preclinical and translational studies
(15). Thus, combining molecular
imaging and gene therapy is likely to be conducive in enhancing the
efficacy and safety of the current gene therapy protocols for human
application and in supporting future individualized patient
treatment (16). In addition, stem
cell studies may require the coexpression of two marker genes, one
driven by a constitutive promoter to monitor gene transfer or track
the modified cells and the other driven by a lineage-specific
promoter to monitor stem cell differentiation. In addition, the
transduction efficiency of baculovirus is quite high in mesenchymal
stem cells (17). Therefore, the
dual-promoter recombinant baculovirus vector significantly
facilitates the monitoring of gene delivery in vitro and
in vivo.
With regard to therapy, certain therapeutic
applications require the coexpression of multiple genes in the same
cell. Specific applications may require the generation of several
proteins that function synergistically as part of a network or
produce multiple subunits of a protein that are expressed by
various genes. There is increasing evidence that effective
treatments for specific diseases may demand the expression of
multiple therapeutic proteins that are selected to treat specific
aspects of the disease process. For example, an Ad5 viral vector
coexpressing human thrombopoietin (hTPO) and human NIS proteins in
tumor cells (Ad-CMV-hTPO-T2A-hNIS) enhanced radioiodine uptake and
prolonged radioiodine retention (18). Furthermore, brain-derived
neurotrophic factor (BDNF) and neurotrophin-3 (NT3) coexpression
using a glucocorticoid (GC)-induced bicistronic expression vector
(pGC-BDNF-IRES-NT3) protected apoptotic cells in a cellular injury
model (19). One study generated
2A-harboring cloning vectors that were likely to be useful for
bicistronic or multicistronic expression (11). In another study, a fusion suicide
gene was combined with human telomerase reverse transcriptase
(hTERT)-targeted shRNA in a new combined plasmid to provide an
antitumor effect via the synergistic actions of suicide gene
therapy and the targeting of hTERT through RNAi (20). However, each strategy has its own
properties and shortcomings, as aforementioned. In the present
study, in view of the advantages of the Bac-to-Bac Baculovirus
Expression system, the pFastBac Dual vector was reconstructed with
primary success.
The pFastBac Dual vector contains two multiple
cloning sites to allow the expression of two heterologous genes.
One gene is controlled by the PH promoter and the other by the p10
promoter, resulting in the production of non-fusion proteins.
Molecular cloning technology can be applied to clone interest
gene(s) into the pFastBac Dual vector. An ATG start codon for the
initiation of translation and a stop codon for the termination of
the gene must be contained in the inserts to ensure proper
expression of recombinant proteins. Once the inserts are cloned
into the pFastBac Dual vector, transformation into DH10Bac E.
coli cells is performed to generate a recombinant bacmid, which
is later transfected into Sf9 cells. Thus, a recombinant
baculovirus is generated following transfection. The Bac-to-Bac
Baculovirus Expression system facilitates the rapid and efficient
generation of a recombinant baculovirus.
In the present study, Bac Dual-CMV-EGFP-CMV-GDNF was
successfully produced and the pFastBac Dual vector was demonstrated
to be an efficient gene transfer vector, fulfilling the strategy of
double gene coexpression. Due to highly efficient gene delivery by
baculoviruses, there have been major advances in the application of
baculoviruses in molecular imaging and gene therapy. Previously,
the most commonly used method for baculovirus-mediated gene imaging
was the coinfection approach, where one vector encodes a gene of
interest and an additional vector encodes a reporter gene. Now, the
reconstructed pFastBac Dual vector is a potential alternative for
gene imaging. In addition, the applications of the pFastBac Dual
vector may be markedly expanded in gene therapy, particularly in
cases requiring dual gene-coordination.
In conclusion, the pFastBac Dual vector with two
promoters of opposite directions was an efficient gene transfer
vector that coexpressed two target genes and served as a platform
for combining reporter or/and therapy genes. Therefore, a novel and
feasible method of coexpression with dual-promoters has been
proposed in the present study. The pFastBac Dual vector is likely
to yield numerous advantages in the future.
Acknowledgements
The study was supported by a grant from the Science
and Technology Commission of Shanghai Municipality (no.
114119a6400).
References
|
1
|
Ehrhardt A, Haase R, Schepers A, Deutsch
MJ, Lipps HJ and Baiker A: Episomal vectors for gene therapy. Curr
Gene Ther. 8:147–161. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen WJ, Xiong ZA, Tang Y, Dong PT, Li P
and Wang ZG: Feasibility and effect of ultrasound
microbubble-mediated wild-type p53 gene transfection of HeLa cells.
Exp Ther Med. 3:999–1004. 2012.PubMed/NCBI
|
|
3
|
Hu YC: Baculoviral vectors for gene
delivery: a review. Curr Gene Ther. 8:54–65. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Guide to Baculovirus Expression Vector
Systemts (BEVS) and Insect Cell Culture Techiniques [Instruction
Manual] Carlsbad, CA, USA, Invitrogen Life Technologies.
|
|
5
|
Hofmann C, Sandig V, Jennings G, Rudolph
M, Schlag P and Strauss M: Efficient gene transfer into human
hepatocytes by baculovirus vectors. Proc Natl Acad Sci USA.
92:10099–10103. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen CY, Lin CY, Chen GY and Hu YC:
Baculovirus as a gene delivery vector: recent understandings of
molecular alterations in transduced cells and latest applications.
Biotechnol Adv. 29:618–631. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ciccarone VC, Polayes DA and Luckow VA:
Generation of recombinant baculovirus DNA in E. coli using a
baculovirus shuttle vector. Methods Mol Med. 13:213–235.
1998.PubMed/NCBI
|
|
8
|
Wang J, Liu S, Wang J, Zhang Y, Li B, Cai
C and Wang S: Study on molecular imaging and radionuclide therapy
of human nasopharyngeal carcinoma cells transfected with
baculovirus-mediated sodium/iodine symporter gene. Int J Oncol.
43:177–184. 2013.
|
|
9
|
Li D and Wang M: Construction of a
bicistronic vector for the co-expression of two genes in
Caenorhabditis elegans using a newly identified IRES.
Biotechniques. 52:173–176. 2012.PubMed/NCBI
|
|
10
|
Blasberg RG: Molecular imaging and cancer.
Mol Cancer Ther. 2:335–343. 2003.
|
|
11
|
Kim JH, Lee SR, Li LH, et al: High
cleavage efficiency of a 2A peptide derived from porcine
teschovirus-1 in human cell lines, zebrafish and mice. PLoS One.
6:e185562011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Björklund A, Kirik D, Rosenblad C,
Georgievska B, Lundberg C and Mandel RJ: Towards a neuroprotective
gene therapy for Parkinson’s disease: use of adenovirus, AAV and
lentivirus vectors for gene transfer of GDNF to the nigrostriatal
system in the rat Parkinson model. Brain Res. 886:82–98. 2000.
|
|
13
|
Airaksinen MS and Saarma M: The GDNF
family: signalling, biological functions and therapeutic value. Nat
Rev Neurosci. 3:383–394. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Piccinini E, Kalkkinen N, Saarma M and
Runeberg-Roos P: Glial cell line-derived neurotrophic factor:
characterization of mammalian posttranslational modifications. Ann
Med. 45:66–73. 2013. View Article : Google Scholar
|
|
15
|
Penheiter AR, Russell SJ and Carlson SK:
The sodium iodide symporter (NIS) as an imaging reporter for gene,
viral, and cell-based therapies. Curr Gene Ther. 12:33–47. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Waerzeggers Y, Monfared P, Viel T,
Winkeler A, Voges J and Jacobs AH: Methods to monitor gene therapy
with molecular imaging. Methods. 48:146–160. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ho YC, Chung YC, Hwang SM, Wang KC and Hu
YC: Transgene expression and differentiation of
baculovirus-transduced human mesenchymal stem cells. J Gene Med.
7:860–868. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li G, Xiang L, Yang W, Wang Z, Wang J and
Chen K: Efficient multicistronic co-expression of hNIS and hTPO in
prostate cancer cells for nonthyroidal tumor radioiodine therapy.
Am J Nucl Med Mol Imaging. 2:483–498. 2012.PubMed/NCBI
|
|
19
|
Wang Y, Gu J, Wang J, et al: BDNF and NT-3
expression by using glucocorticoid-induced bicistronic expression
vector pGC-BDNF-IRES-NT3 protects apoptotic cells in a cellular
injury model. Brain Res. 1448:137–143. 2012. View Article : Google Scholar
|
|
20
|
Li J, Zhang G, Liu T, Gu H, Yan L and Chen
B: Construction of a novel vector expressing the fusion suicide
gene yCDglyTK and hTERT-shRNA and its antitumor effects. Exp Ther
Med. 4:442–448. 2012.PubMed/NCBI
|