Introduction
Chemotactic factors are a class of cytokines that
cause the migration of special receptor-expressing cells, playing
an important role in the inflammatory reaction. Chemokine ligand 2
(CCL2), which was previously known as monocyte chemoattractant
protein-1, is a monocyte chemotactic factor. CCL2 can specifically
bind to its receptor, chemokine receptor type 2 (CCR2), to regulate
the migration and infiltration of monocytes, T cells and natural
killer cells in the inflammatory area (1–3).
CCR2, a G protein-coupled receptor, is widely expressed on
endothelial cells, horizontal cells, gitter cells and neurons in
the central nervous systems of a number of species in addition to
humans (4–7). Furthermore, CCL2 and CCR2 have been
demonstrated to be expressed in numerous encephalic regions in
addition to the hippocampus (6–8). The
expression pattern of CCR2 can be altered in a neuropathological
state.
Cerebral ischemia is often accompanied with
reperfusion injury, thus, therapy targeted to reduce the damage is
urgently required in clinical practice. Since there is no
clinically effective method to treat cerebral ischemia/reperfusion
injury to date, it is necessary to further study this disease. The
selection of anesthetic drugs for patients with cerebral ischemia
in the perioperative period requires careful consideration by
anesthesiologists. Propofol is a common anesthetic drug used widely
as a clinical anesthesia due to the numerous advantages, including
rapid action, quick clearance and fewer adverse side effects.
A number of studies have demonstrated that propofol
exhibits protective roles in the brain, however, the majority of
studies have been conducted with ischemia models (9,10).
Propofol has also been reported to exhibit a potential protective
role in procerebral ischemia/reperfusion injury (11), however, the exact mechanism remains
unclear. In the present study, the effects of preadministration of
propofol on the expression levels of CCL2 and CCR2 in the
hippocampus were investigated in rats with procerebral
ischemia/reperfusion injury. The aim of the study was to provide
more useful evidence for the further study of cerebral
ischemia/reperfusion injury.
Materials and methods
Animals and grouping
In total, 24 healthy adult male Sprague-Dawley rats,
weighing 250–300 g, were obtained from the Physiological Laboratory
Animal Center at Shanxi Medical University (Taiyuan, China). The
study was conducted in strict accordance with the recommendations
in the Guide for the Care and Use of Laboratory Animals of the
National Institutes of Health, and the animal use protocol was
reviewed and approved by the Institutional Animal Care and Use
Committee of Shanxi Provincial People’s Hospital (Taiyuan, China).
Animals were housed in a specific pathogen-free room at a constant
temperature of 25°C and a relative humidity of 45%. All the animals
received humane care in compliance with the strict guiding
principles of the National Institution of Health for Experimental
Care and Use of Animals. The experimental design and procedures
were approved by the Institutional Ethical Committee for Animal
Care and Use of Shanxi Provincial People’s Hospital.
Rats were randomly divided into three groups (n=8):
Sham-operated (C group), cerebral ischemia/reperfusion injury (I/R
group) and propofol-intervention groups (P group). Rats in the
sham-operated group received all the surgical procedures, but
without the bloodletting and reinfusing process. In the P group, a
dosage of 50 mg/kg propofol (FJ261, AstraZeneca, Milan, Italy) was
injected into the vena femoralis of the rats prior to the cerebral
ischemia surgery. Rats in the I/R group were administered 1 ml
physiological saline instead of propofol by vena femoralis
injection prior to the cerebral ischemia surgery.
I/R protocol
Rats were anesthetized with 10% chloral hydrate (0.3
ml/kg, i.p.) and fixed in a supine position. Following a median
incision of the neck skin, the bilateral common carotid artery was
carefully dissected and exposed. The left femoral artery and vein
were cannulated with NO.24 trochar. The arterial blood pressure was
monitored continuously using a BL-420F Bio-function Experimental
System (Thai Meng Technology Co., Ltd., Chengdu, China). At 15 min
prior to bloodletting, the venous blood gas was measured
periodically (AVL-939 mini blood gas pH analyzer; Switzerland) to
maintain the pH, arterial carbon dioxide tension (PaCO2)
and arterial oxygen tension (PaO2) at normal ranges (pH,
7.35–7.45; PaCO2, 35–45 mmHg; PaO2, 290–140
mmHg), where 1 kPa is equal to 7.5 mmHg. Next, the rats were bled
slowly over 5 min from the left femoral vein by withdrawing or
infusing blood into a 10 ml heparinized syringe to maintain the
mean arterial pressure between 35 and 45 mmHg. Immediately after
reaching 35 mmHg, the common carotid arteries were occluded using
atraumatic aneurysm clamps for 10 min to induce procerebral
ischemia. The brain circulation was restored by unclamping the
common carotid arteries and reinfusing the blood into the rat for 5
min. The trochars were removed from the femoral artery and vein to
terminate the reperfusion.
Sampling
Rats were sacrificed by decapitation at 6 h after
reperfusion and anesthesia. The brain was removed and the bilateral
hippocampus was separated immediately on plates at −20°C. The
hippocampi collected were frozen quickly in liquid nitrogen and
stored at −80°C.
Quantitative polymerase chain reaction
(qPCR)
For qPCR, 50-mg samples of hippocampal tissue were
disrupted in 1 ml TRIzol reagent (Invitrogen Life Technologies,
Carlsbad, CA, USA) to form a homogenate. Next, the homogenates were
treated with chloroform, isopropyl alcohol and ethanol to extract
the total RNA. cDNA synthesis was conducted on the RNA product
using a PrimeScript RT reagents kit (Takara Bio, Inc., Shiga,
Japan), according to the manufacturer’s instructions. PCR was
performed in a total volume of 50 μl (containing 3 μl cDNA, 50 pmol
forward and reverse primers, 0.4 μmol/l dNTP, 5 μl 10X buffer and
2.5 IU Taq polymerase) using a Gradient PCR machine
(Biometra GmbH, Göettingen, Germany). The following gene-specific
primer pairs were used: CCL2, forward, 5′-CTG TCT CAG CCA GAT GCA
GTT-3′ and reverse, 5′-GAG CTT GGT GAC AAA TAC TAC A-3′; CCR2
forward, 5′-GTT CTC TTC CTG ACC ACC TTC-3′ and reverse, 5′-CTT CGG
AAC TTC TCA CCA ACA-3′; β-actin forward, 5′-TCC CTC AAG ATT GTC AGC
AA-3′ and reverse, 5′-AGA TCC ACA ACG GAT ACA TT-3′. The product
sizes were 147, 157 and 308 bp, respectively. All the samples were
run in cycles as follows: One cycle of 94°C for 3 min; 35 cycles of
94°C for 30 sec, 50°C for 30 sec and 72°C for 1 min; one cycle of
72°C for 5 min. The samples were then cooled to 4°C.
The final PCR products were run on 2% agarose gel,
stained with ethidium bromide and observed using a viltalight lamp.
A DNA marker with an 100-bp ladder was used to identify the product
sizes. The stained DNA bands were then analyzed with a computer gel
image analysis system (Kodak, Rochester, NY, USA). The density of
the PCR products was calculated using the area under the curves,
while the relative mRNA expression levels of CCL2 and CCR2 were
determined using the ratio of the density of CCL2 or CCR2 against
that of β-actin.
Western blot analysis
Total protein of the hippocampal tissue was
extracted using a protein extraction kit (Apply Gen Technologies,
Inc., Beijing, China). The proteins were separated with SDS-PAGE
and transferred onto polyvinylidene fluoride membranes for
immunoblotting. Next, 10% defatted milk powder dissolved in
Tris-buffered saline Tween-20 (TBST) was added to the membranes to
block the endogenous horseradish peroxidase for 1 h in room
temperature. The membranes were then incubated with mouse anti-rat
CCL2, CCR2 and β-actin antibodies (1:1,000; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) overnight at 4°C.
Following washing with TBST three times, each time for 10 min, the
membranes were incubated with horseradish peroxidase-conjugated
anti-mouse IgG (1:10,000; Beijing Zhongshan Golden Bridge
Biotechnology Co., Ltd., Beijing, China) for 2 h at room
temperature. The specific protein bands on the membranes were
visualized using an enhanced chemiluminescence kit, according to
the manufacturer’s instructions. The bands were scanned using a
Gel-imaging system (Bio-Rad 2000; Bio-Rad, Hercules, CA, USA) and
analyzed using Quantity One software. The optical density values of
the two targeted proteins were calibrated against that of β-actin,
from which the protein expression levels of CCL2 and CCR2 were
calculated.
Statistical analysis
Statistical analysis was performed with the SPSS
software program v 11.0 (SPSS, Inc., Chicago, IL, USA). Data are
expressed as the mean ± standard deviation. Differences among the
groups were compared using one-factor analysis of variance, where
P<0.05 was considered to indicate a statistically significant
difference.
Results
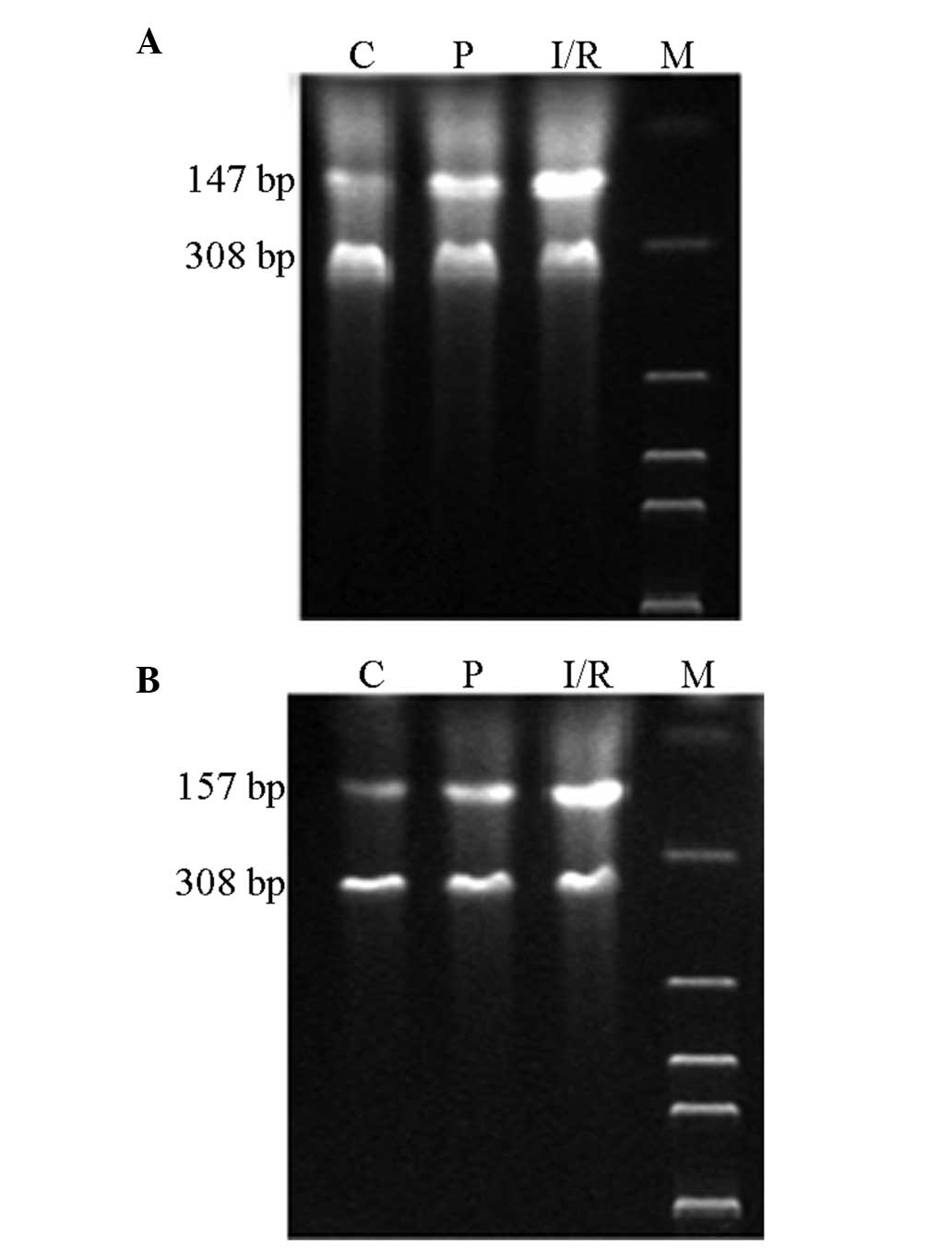
Relative mRNA expression levels of CCL2
and CCR2 in rats
Expression levels of CCL2 and CCR2 mRNA in the
hippocampus were analyzed by qPCR following cerebral
ischemia/reperfusion. The mRNA expression levels of CCL2 and CCR2
in the hippocampus neurons of the I/R and P groups increased
significantly (P<0.05) when compared with the expression levels
in the C group. However, the expression levels of CCL2 and CCR2 in
the hippocampus neurons of the P group decreased markedly
(P<0.05) when compared with the I/R group (Table I; Fig.
1).
 | Table IRelative mRNA expression levels of
CCL2 and CCR2. |
Table I
Relative mRNA expression levels of
CCL2 and CCR2.
| Group | CCL2 | CCR2 |
|---|
| C | 0.49±0.27 | 0.29±0.13 |
| I/R | 1.58±0.42a | 0.56±0.21a |
| P | 0.76±0.29b | 0.47±0.22b |
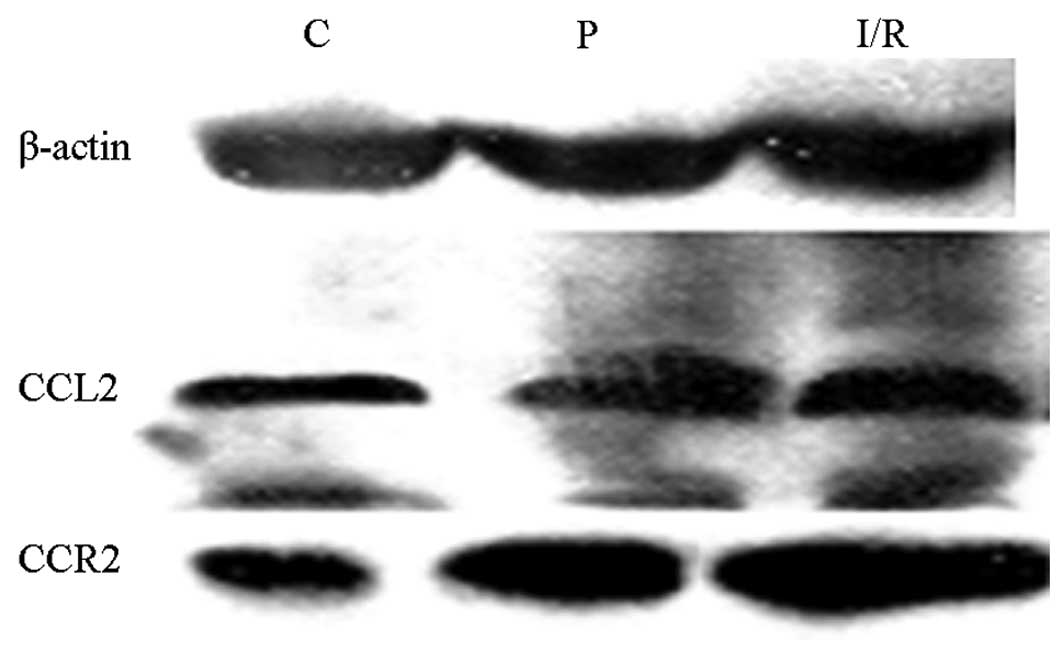
Protein expression levels of CCL2 and
CCR2 in rats
Protein expression levels of CCL2 and CCR2 in the
hippocampus were analyzed by western blot analysis following
cerebral ischemia/reperfusion. The protein expression levels of
CCL2 and CCR2, as detected by western blot analysis, are shown in
Fig. 2. Expression levels of CCL2
and CCR2 in the I/R and P groups were significantly higher compared
with those in the C group. In addition, CCL2 and CCR2 expression
levels in the P group decreased markedly when compared with those
in the I/R group (Table II).
| Table IIProtein expression levels of CCL2 and
CCR2. |
Table II
Protein expression levels of CCL2 and
CCR2.
| Group | CCL2 | CCR2 |
|---|
| C | 0.21±0.016 | 0.23±0.023 |
| I/R | 0.79±0.072a | 0.47±0.085a |
| P | 0.56±0.029b | 0.33±0.069b |
Discussion
Preliminary investigations demonstrated that the
mRNA expression levels of CCL2 in the brain cortex increased
significantly at 2 h after ischemia in an ischemia/reperfusion
model. Expression levels peaked at 16 h after reperfusion and were
maintained at a high level for 48 h following reperfusion.
Combining the results from the preliminary experiments, in the
present study, the expression levels of CCL2 and CCR2 were
investigated in rat hippocampal neurons at 6 h after reperfusion.
In addition, the present study investigated the protective roles of
propofol on the brain by injecting propofol into the vena femoralis
prior to ischemia surgery.
The neuroprotective effect of propofol is the result
of a number of mechanisms, including reducing the brain oxygen
metabolism rate, removing the oxygen free radicals (12,13),
activating the c-aminobutyric acid type A receptor (14), inhibiting the glutamate receptors
(15), reducing the extracellular
glutamate concentration (16) and
increasing the glutamic acid salt intake (17) via inhibiting the release of
glutamate-dependent Na+ channels. In the present study,
propofol was shown to significantly downregulate the expression
levels of CCL2 and CCR2 in the hippocampal tissues, indicating that
propofol may decrease the inflammatory reactions caused by ischemia
during reperfusion. Propofol may exhibit this neuroprotective
effect in cerebral ischemia/reperfusion injury by regulating the
expression levels of CCL2 and CCR2.
Clinically, anesthetic drugs are often administered
prior to the occurrence of brain ischemia. Models can imitate
cerebral ischemia caused by acute hemorrhage, cardiac arrest and
certain shock (18). Therefore,
future research on the mechanisms underlying hypoxic-ischemic brain
damage should investigate the effects of CCL2 and CCR2 using rats
with cerebral ischemia/reperfusion.
Previous research with regard to stroke has also
demonstrated that overexpression of CCL2 can aggravate ischemic
brain injury, and inhibiting the expression of CCL2 can reduce
injury. Inflammatory reactions are known to play important roles in
brain injury (19). In axon
injury, the CCL2 that is produced by early glial cells causes an
intrinsic reaction, inducing leukocyte infiltration. In addition,
CCL2 can supplement T-cell activation-3/CCL1, the ligand of the
CCR8 receptor, to guide the infiltration of T cells in
encephalomyelitis (20). Thus,
chemotactic factors derived from glial cells can be used as key
regulators to accommodate the central immune system (21). However, CCR2 and CCL2 are critical
for the attraction of leukocyte infiltration (22), and the interaction between them
also influences the processing and clearance of cerebral hemorrhage
(23–24).
CCR2 is widely expressed on brain neurons,
astrocytes and gitter cells, and is a high-affinity receptor for
CCL2. When CCL2 binds to its receptor, monocytes are caused to
migrate to the inflammatory sites and participate in the
inflammatory reactions mediated by monocytes. The results of the
present study indicated that there was a synchronous increase in
the expression levels of CCL2 and CCR2 in cerebral
ischemia/reperfusion injury and a synchronous decrease when
propofol was injected into the rats prior to surgery. These
observations indicate that preadministration of propofol may
suppress the inflammatory reaction by regulating the expression
levels of CCL2 and CCR2, which then consequently reduces the danger
of procerebral ischemia/reperfusion injury in rats.
However, only one time point and single dosage were
analyzed in the present study. Data with more time points and
multiple administration concentrations, as well as morphological
evidence, may supplement these results in the future.
In conclusion, CCL2 and CCR2 are involved in the
pathogenic mechanisms underlying cerebral ischemia/reperfusion
injury in rats, and preadministration of propofol can suppress the
expression of CCL2 and CCR2. In-depth study to investigate the
exact roles of chemotactic factors in immunological injury of the
central nervous system may be of great importance to improve the
prognosis of cerebral ischemia/reperfusion injury and identify
specific novel therapeutic targets.
References
|
1
|
Silbernagel G, Machann J, Häring HU,
Fritsche A and Peter A: Plasminogen activator inhibitor-1, monocyte
chemoattractant protein-1, e-selectin and C-reactive protein levels
in response to 4-week very-high-fructose or -glucose diets. Eur J
Clin Nutr. 68:97–100. 2014. View Article : Google Scholar
|
|
2
|
Hazalin NA, Lim SM, Cole AL, Majeed AB and
Ramasamy K: Apoptosis induced by desmethyl-lasiodiplodin is
associated with upregulation of apoptotic genes and downregulation
of monocyte chemotactic protein-3. Anticancer Drugs. 24:852–861.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shimizu S, Nakashima H, Karube K, Ohshima
K and Egashira K: Monocyte chemoattractant protein-1 activates a
regional Th1 immunoresponse in nephritis of MRL/lpr mice. Clin Exp
Rheumatol. 23:239–242. 2005.PubMed/NCBI
|
|
4
|
Chaturvedi LS, Zhang P and Basson MD:
Effects of extracellular pressure and alcohol on the microglial
response to inflammatory stimulation. Am J Surg. 204:602–606. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Volpe S, Cameroni E, Moepps B, et al: CCR2
acts as scavenger for CCL2 during monocyte chemotaxis. PLoS One.
7:e372082012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Banisadr G, Gosselin RD, Mechighel P, et
al: Constitutive neuronal expression of CCR2 chemokine receptor and
its colocalization with neurotransmitters in normal rat brain:
functional effect of MCP-1/CCL2 on calcium mobilization in primary
cultured neurons. J Comp Neurol. 492:178–192. 2005. View Article : Google Scholar
|
|
7
|
Banisadr G, Gosselin RD, Mechighel P, et
al: Highly regionalized neuronal expression of monocyte
chemoattractant protein-1 (MCP-1/CCL2) in rat brain: evidence for
its colocalization with neurotransmitters and neuropeptides. J Comp
Neurol. 489:275–292. 2005. View Article : Google Scholar
|
|
8
|
Foresti ML, Arisi GM, Katki K, et al:
Chemokine CCL2 and its receptor CCR2 are increased in the
hippocampus following pilocarpine-induced status epilepticus. J
Neuroinflammation. 6:402009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Harman F, Hasturk AE, Yaman M, et al:
Neuroprotective effects of propofol, thiopental, etomidate, and
midazolam in fetal rat brain in ischemia-reperfusion model. Childs
Nerv Syst. 28:1055–1062. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
McIntosh MP and Rajewski RA: Comparative
canine pharmacokinetics-pharmacodynamics of fospropofol disodium
injection, propofol emulsion, and cyclodextrin-enabled propofol
solution following bolus parenteral administration. J Pharm Sci.
101:3547–3552. 2012. View Article : Google Scholar
|
|
11
|
Lee Y, Chung C and Oh YS: Effectiveness of
propofol pretreatment on the extent of deranged cerebral
mitochondrial oxidative enzyme system after incomplete forbrain
ischemia/reperfusion in rats. J Korean Med Sci. 15:627–630. 2000.
View Article : Google Scholar
|
|
12
|
Peters CE, Korcok J, Gelb AW and Wilson
JX: Anesthetic concentrations of propofol protect against oxidative
stress in primary astrocyte cultures: comparison with hypothermia.
Anesthesiology. 94:313–321. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tsuchiya M, Asada A, Maeda K, et al:
Propofol versus midazolam regarding their antioxidant activities.
Am J Respir Crit Care Med. 163:26–31. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Buggy DJ, Nicol B, Rowbotham DJ and
Lambert DG: Effects of intravenous anesthetic agents on glutamate
release: a role for GABAA receptor-mediated inhibition.
Anesthesiology. 92:1067–1073. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu D, Jiang Y, Gao J, Liu B and Chen P:
Repeated exposure to propofol potentiates neuroapoptosis and
long-term behavioral deficits in neonatal rats. Neurosci Lett.
534:41–46. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kobayashi M, Takeda Y, Taninishi H, et al:
Quantitative evaluation of the neuroprotective effects of
thiopental sodium, propofol, and halothane on brain ischemia in the
gerbil: effects of the anesthetics on ischemic depolarization and
extracellular glutamate concentration. J Neurosurg Anesthesiol.
19:171–178. 2007. View Article : Google Scholar
|
|
17
|
Cai J, Hu Y, Li W, et al: The
neuroprotective effect of propofol against brain ischemia mediated
by the glutamatergic signaling pathway in rats. Neurochem Res.
36:1724–1731. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li J, Han B, Ma X and Qi S: The effects of
propofol on hippocampal caspase-3 and Bcl-2 expression following
forebrain ischemia-reperfusion in rats. Brain Res. 1356:11–23.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ji K and Tsirka SE: Inflammation modulates
expression of laminin in the central nervous system following
ischemic injury. J Neuroinflammation. 9:1592012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Murphy CA, Hoek RM, Wiekowski MT, Lira SA
and Sedgwick JD: Interactions between hemopoietically derived TNF
and central nervous system-resident glial chemokines underlie
initiation of autoimmune inflammation in the brain. J Immunol.
169:7054–7062. 2002. View Article : Google Scholar
|
|
21
|
Babcock AA, Kuziel WA, Rivest S and Owens
T: Chemokine expression by glial cells directs leukocytes to sites
of axonal injury in the CNS. J Neurosci. 23:7922–7930.
2003.PubMed/NCBI
|
|
22
|
Yao Y and Tsirka SE: The CCL2-CCR2 system
affects the progression and clearance of intracerebral hemorrhage.
Glia. 60:908–918. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Andres RH, Choi R, Pendharkar AV, Gaeta X,
Wang N, et al: The CCR2/CCL2 interaction mediates the
transendothelial recruitment of intravascularly delivered neural
stem cells to the ischemic brain. Stroke. 42:2923–2931. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Semple BD, Kossmann T and
Morganti-Kossmann MC: Role of chemokines in CNS health and
pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J
Cereb Blood Flow Metab. 30:459–473. 2010. View Article : Google Scholar : PubMed/NCBI
|