Introduction
Type 2 diabetes (T2D) is a major public health
problem in numerous countries (1,2).
Genetic and environmental factors are hypothesized to contribute to
the initiation and development of the disease (3). Genetic association studies based on
genome-wide association and meta-analyses have revealed a number of
susceptible genetic polymorphisms associated with the pathogenesis
of T2D (2,4–7).
However, a number of these polymorphisms are not functional
variants and are most likely cotransmitted with the causative
polymorphisms in large linkage disequilibrium blocks, and do not
directly control the expression of T2D susceptibility genes
(8). Evaluating the impact of
these genetic-environmental interactions and predicting the
personal onset risk of developing T2D remains a challenge.
Epigenetic regulation is sensitive to a number of
environmental factors and may provide a bridge between the
environment and genetic background (1). As a crucial mechanism in epigenetic
regulation, DNA methylation regulates gene expression by
transferring a methyl group to cytosine nucleotides via DNA
methyltransferase (1). Unlike
genetic polymorphisms, DNA methylation levels may alter during
numerous processes, including development, tissue differentiation
and aging (1). In a monozygotic
twins study, the older twins showed more distinct changes according
to DNA methylation profiles in the epigenome (9,10).
Environmental factors, including nutrition and lifestyle, may
influence DNA methylation in mammals. For example, the promoter
methylation of certain genes was increased in human primary muscle
cells due to the exposure of the free fatty acids palmitate
(10). A series of genes in
primary metabolic processes were reported to show a differential
methylation status in skeletal muscle in patients with T2D compared
with normal glucose-tolerant individuals (11). Furthermore, dysregulation of
promoter methylation has been found in pancreatic islets from
patients with T2D (1). Elucidating
the association between promoter methylation and the pathogenesis
of T2D is a promising field in the research of metabolic disorders
(1,10).
The calcium/calmodulin-dependent protein kinase 1D
(CAMK1D) gene encodes a member of the
Ca2+/calmodulin-dependent protein kinase 1 subfamily of
serine/threonine kinases that plays an important role in the
regulation of granulocyte function via the chemokine signaling
transduction pathway (12,13). Single nucleotide polymorphisms
(SNPs) in CAMK1D have been found to be associated with the
susceptibility of developing T2D in an east Asian population
(14). However, no significant
association between the CAMK1D SNP and T2D was found in a study
based on European populations (15). Thus, the role of the CAMK1D gene in
the pathophysiology of T2D remains unclear (13). The calmodulin 2 (CALM2) gene
encodes a member of the human calmodulin family, and polymorphisms
in this gene have been associated with dialysis survival in
T2D-associated renal disease (16). The cryptochrome 2 (CRY2) gene is a
circadian signal gene, whose genetic variation has been associated
with T2D and metabolic characteristics (17,18).
A recent study investigating monozygotic twins found that the
methylation status of CRY2 in subcutaneous adipose tissue differed
between the twin with T2D and the healthy twin (18). Further investigations into the
association between promoter methylation of the CRY2 gene and T2D
in the peripheral blood should be conducted.
Aberrant gene methylation has been identified not
only in diseased tissues, but also in human peripheral tissue,
including blood lymph cells (8).
Detecting the promoter methylation levels of T2D-associated genes
in the peripheral blood may be beneficial for the identification of
novel diabetic biomarkers with preventative and/or diagnostic
values. In the present study, the methylation levels of three
candidate genes (CAMK1D, CRY2 and CALM2) in patients with T2D and
non-diabetic individuals were investigated to determine the value
of this epigenetic marker, with the aim of providing further
understanding into the disease etiology of T2D.
Materials and methods
Subjects
A total of 48 patients with T2D and 48 age- and
gender-matched healthy controls were recruited from the Affiliated
Hospital of Ningbo University (Ningbo, China). The characteristics
of the individuals are shown in Table
I. All the individuals were of Han Chinese origin and had lived
in the Ningbo area for at least three generations. Patients with
T2D were recruited if their plasma glucose levels were >7.0
mmol/l at fasting or >11.1 mmol/l at 2 h following glucose load
(World Health Organization) (19).
Healthy individuals were recruited according to the standard of
fasting blood glucose of <6.1 mmol/l. None of the controls had a
family history of T2D in first-degree relatives or had received any
medication. Subjects were excluded from the study if they had
hypertension, coronary heart disease, renal inadequacy, drug abuse
or any other serious diseases. The study was approved by the Ethics
Committee of Ningbo University and written informed consent was
obtained from all the subjects. Blood samples were collected in
3.2% citrate sodium-treated tubes and stored at −80°C for DNA
extraction.
 | Table ICharacteristics of the subjects
(n=96). |
Table I
Characteristics of the subjects
(n=96).
| Characteristics | Value | Range | T2D | Controls | P-value |
|---|
| Age (years) | 59.2±7.5 | 35–69 | 59.2±7.5 | 59.2±7.5 | 1.000 |
| Gender (M/F) | 48/48 | - | - | - | - |
| BMI
(kg/m2) | 23.71±3.28 | 17.15–42.96 | 24.17±4.18 | 23.18±1.64 | 0.146 |
| Total cholesterol
(mmol/l) | 5.19±0.96 | 2.95–7.90 | 5.34±0.83 | 5.05±1.06 | 0.140 |
| Total triglycerides
(mmol/l) | 1.60±1.36 | 0.40–9.92 | 1.90±1.69 | 1.31±0.82 | 0.034 |
| Glucose (mmol/l) | 6.76±2.65 | 4.38–22.84 | 8.31±2.91 | 5.22±0.92 | 0.000 |
| ALT (IU/l) | 21.5±15.9 | 5.0–99.0 | 25.1±18.5 | 18.0±12.1 | 0.028 |
| Uric acid
(μmol/l) | 294.9±81.1 | 132.0–531.0 | 289.3±70.5 | 300.6±90.9 | 0.499 |
| CALM2 methylation
(%) |
| CpG1 | 0.96±0.38 | 0–2 | 1.02±0.40 | 0.88±0.33 | 0.099 |
| CpG2 | 2.01±0.50 | 0–3 | 2.18±0.39 | 1.79±0.55 | 0.001 |
| CpG3 | 0.92±0.48 | 0–2 | 1.00±0.53 | 0.82±0.39 | 0.087 |
| CpG4 | 1.40±0.86 | 0–5 | 1.11±0.58 | 1.79±1.02 | 0.001 |
| CRY2 methylation
(%) |
| CpG1 | 1.25±0.53 | 0–2 | 1.13±0.41 | 1.44±0.65 | 0.022 |
| CpG2 | 0.80±0.41 | 0–1 | 0.79±0.41 | 0.80±0.41 | 0.961 |
| CpG3 | 1.61±0.63 | 0–3 | 1.64±0.63 | 1.56±0.65 | 0.621 |
| CpG4 | 0.98±0.42 | 0–3 | 1.05±0.39 | 0.88±0.44 | 0.110 |
| CpG5 | 1.14±3.32 | 0–27 | 0.64±0.54 | 1.92±5.24 | 0.134 |
| CAMK1D methylation
(%) |
| CpG1 | 1.04±0.29 | 0–3 | 1.04±0.20 | 1.04±0.36 | 1.000 |
| CpG2 | 0.75±0.48 | 0–2 | 0.90±0.31 | 0.60±0.57 | 0.003 |
| CpG3 | 1.53±0.54 | 1–3 | 1.75±0.48 | 1.31±0.51 | 0.000 |
| CpG4 | 1.04±0.29 | 0–2 | 1.10±0.31 | 0.98±0.25 | 0.032 |
| CpG5 | 1.02±0.20 | 0–2 | 1.04±0.20 | 1.00±0.21 | 0.320 |
| CpG6 | 0.96±0.32 | 0–2 | 1.02±0.14 | 0.90±0.43 | 0.057 |
| CpG7 | 1.04±0.29 | 0–2 | 1.06±0.38 | 1.02±0.14 | 0.480 |
| CpG8 | 0.92±0.45 | 0–3 | 1.02±0.25 | 0.81±0.57 | 0.023 |
| CpG9 | 0.82±0.54 | 0–3 | 1.00±0.41 | 0.65±0.60 | 0.001 |
Phenotype collection
Blood samples were collected from the antecubital
vein into vacutainer tubes containing ethylene diamine tetraacetic
acid following fasting for 12 h overnight. Plasma levels of
cholesterol, triglyceride, alanine aminotransferase (ALT), uric
acid and glucose concentrations were enzymatically measured using
the CX7 Analyzer (Beckman Coulter, Inc., Fullerton, CA, USA).
DNA methylation assay
Human genomic DNA was isolated from peripheral blood
samples using a nucleic acid extraction automatic analyzer (Lab-Aid
820; Xiamen, Fujian, China). DNA was quantified using the PicoGreen
double strand DNA Quantification kit (Molecular Probes, Inc.,
Eugene, OR, USA). Bisulfite pyrosequencing technology was used to
determine the CpG dinucleotide methylation levels of fragments
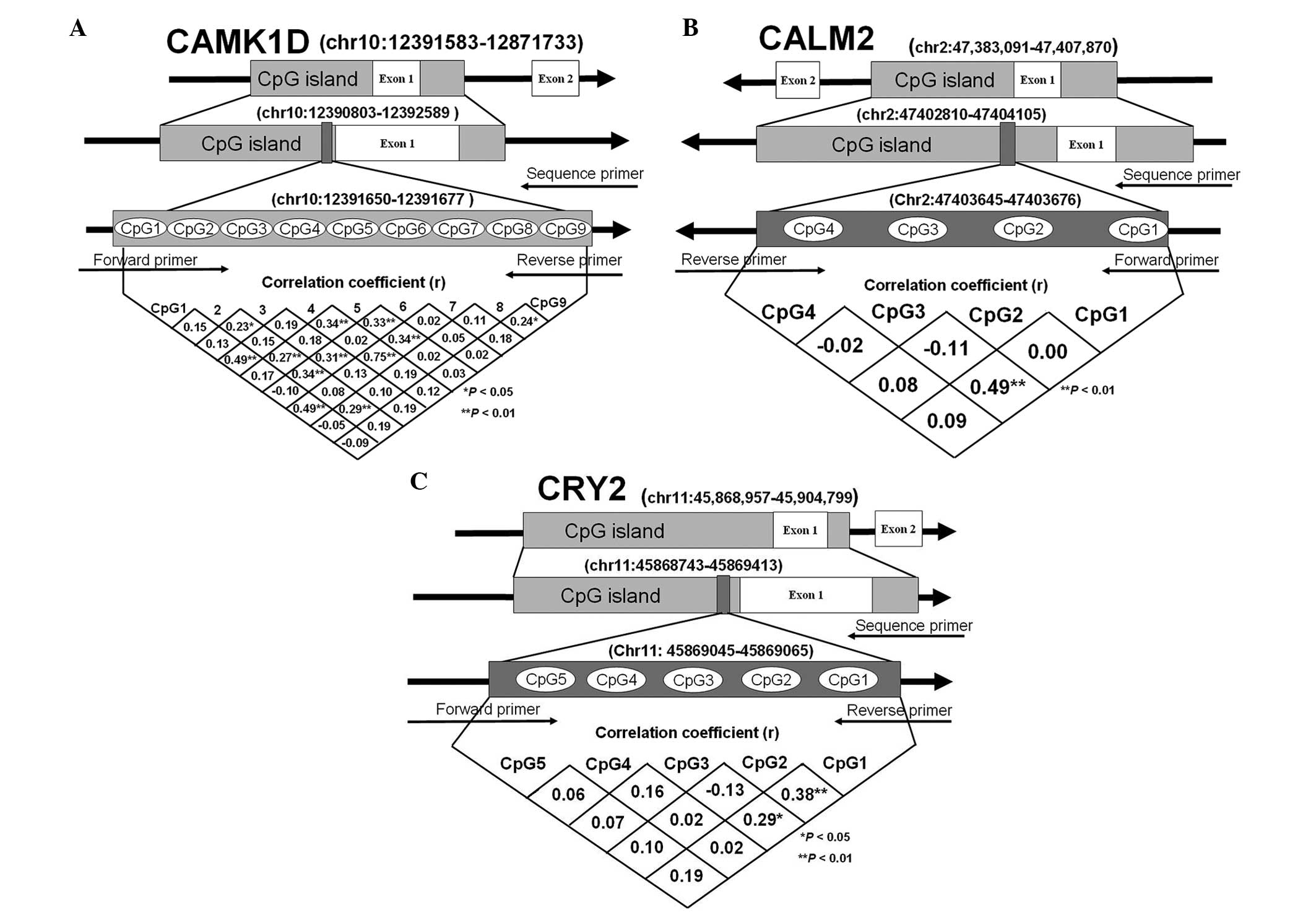
within the promoters of the CAMK1D, CRY2 and CALM2 genes (Fig. 1). The pyrosequencing assays
involved sodium bisulfite DNA conversion chemistry, polymerase
chain reaction (PCR) amplification and sequencing by synthesis
assay of the target sequence. Sodium bisulfite preferentially
deaminates unmethylated cytosine residues to thymines (following
PCR amplification), while methyl-cytosines remain unmodified. PCR
primers were selected using PyroMark Assay Design software
v2.0.1.15 and the amplification primers for the CAMK1D, CRY2 and
CALM2 gene promoters are shown in Table II.
| Table IIPrimers for the promoter CpG
methylation analysis. |
Table II
Primers for the promoter CpG
methylation analysis.
| Gene | Sequence |
|---|
| CALM2 |
| Forward
primer |
5′-AGGAGGAGTTGTTGGAGAATATGA-3′ |
| Reverse
primer | 5′-biotin-
ACTACCCCCCTAACCCCTCT-3′ |
| Sequencing
primer |
5′-GTTTTGAGTGTTTAGGTAAGG-3′ |
| CRY2 |
| Forward
primer |
5′-GGGGTGGTTGGAGTAGTTTGG-3′ |
| Reverse
primer |
5′-biotin-AATCCCCTCACCTCCATC-3′ |
| Sequencing
primer |
5′-GGAGTAGTTTGGATAGTTA-3′ |
| CAMK1D |
| Forward
primer |
5′-GGAGGTAAGAAAGTAGTAGAAAGTGA-3′ |
| Reverse
primer |
5′-biotin-CCTCCTCTACAATTTCCTCTT-3′ |
| Sequencing
primer |
5′-GAGTTAGGGAGGGAT-3′ |
Statistical analysis
Pearson’s χ2 test was used to compare the
categorical variables, while mean group differences for continuous
variables were compared using the Student’s t-test. Pearson’s
correlation analysis was applied to determine the associations
between the methylation status and metabolic features of the
subjects. P<0.05 was considered to indicate a statistically
significant difference. All statistical analyses were performed
using PASW Statistics 18.0 software (SPSS, Inc., Chicago, IL,
USA).
Results
Low methylation levels within the CAMK1D,
CRY2 and CALM2 promoters
A total of 48 patients with T2D and 48 age- and
gender-matched controls were recruited for the association study,
and the methylation levels of three genes (CAMK1D, CRY2 and CALM2)
were investigated. In total, four CpG dinucleotides within the
CALM2 gene promoter, five CpG dinucleotides within the CRY2 gene
promoter and nine CpG dinucleotides within the CAMK1D gene promoter
were identified. The correlations between the DNA methylation
levels among all the CpGs are shown in Fig. 1 and the characteristics of the
subjects are shown in Table I. As
shown in Table I, the promoters of
the three genes in the peripheral blood exhibited low methylation
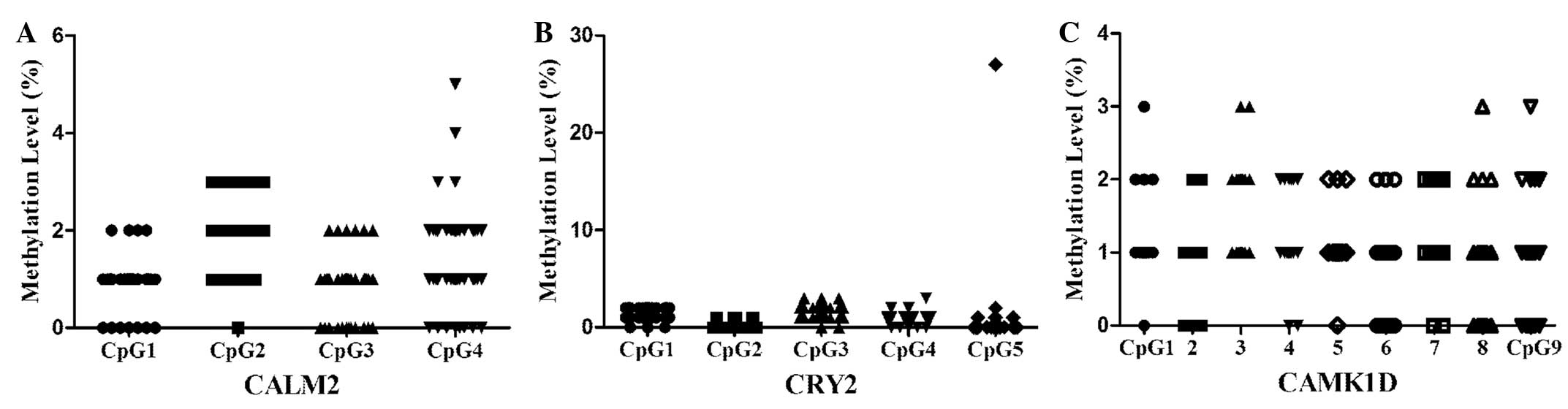
levels for all the subjects. The DNA methylation information of all
the CpGs within the CAMK1D, CRY2 and CALM2 promoters is shown in
Fig. 2.
Statistical analysis of clinical
phenotypes
No statistically significant differences in clinical
phenotypes, including age, body mass index, total cholesterol and
uric acid, were observed between the T2D and controls patients
(Table I; P>0.05). However, the
glucose level in patients with T2D was significantly higher
compared with the control patients (Table I; P<0.001), and a similar result
was observed in the breakdown analysis by gender. The levels of ALT
and total triglycerides were higher in patients with T2D (Table I; P=0.028 and P=0.034,
respectively). However, in the breakdown analysis by gender, no
differential level of total triglycerides was observed. Notably,
higher levels of ALT in female patients with T2D were observed
(Table III; P=0.019).
| Table IIICharacteristics of the subjects by
gender. |
Table III
Characteristics of the subjects by
gender.
|
Characteristics | T2D | Controls | P-value |
|---|
| Males (n=48) |
| Age (years) | 59.1±8.7 | 59.1±8.7 | |
| BMI
(kg/m2)a | 24.92±5.17 | 23.10±1.21 | 0.124 |
| Total cholesterol
(mmol/l) | 5.06±0.74 | 4.86±1.11 | 0.464 |
| Total
triglycerides (mmol/l) | 1.81±1.56 | 1.38±0.90 | 0.254 |
| Glucose
(mmol/l) | 8.59±3.49 | 4.94±0.34 | 0.000 |
| ALT (IU/l) | 30.4±23.8 | 21.1±15.9 | 0.119 |
| Uric acid
(μmol/l) | 304.7±70.6 | 346.5±82.7 | 0.066 |
| Females (n=48) |
| Age (years) | 59.4±6.4 | 59.4±6.4 | |
| BMI
(kg/m2)b | 23.49±2.97 | 23.25±1.93 | 0.747 |
| Total cholesterol
(mmol/l) | 5.62±0.83 | 5.24±1.00 | 0.161 |
| Total
triglycerides (mmol/l) | 1.98±1.85 | 1.23±0.75 | 0.071 |
| Glucose
(mmol/l) | 8.04±2.22 | 5.49±1.20 | 0.000 |
| ALT (IU/l) | 19.8±8.6 | 14.8±4.8 | 0.019 |
| Uric acid
(μmol/l) | 273.9±68.3 | 254.6±75.0 | 0.357 |
Discussion
Epigenetic regulation is involved in numerous types
of human diseases, and epigenetic biomarkers have been reported to
be associated with pathological processes, of which a number have
potential value as laboratory diagnostic tools (20–23).
DNA methylation, as one of the most studied epigenetic regulatory
mechanisms, is essential in the interaction between genetic and
environmental factors (10,20).
DNA methylation usually occurs in the CpG islands of functional
gene promoter regions (20), and
this type of epigenetic modification was originally considered to
be stable in vitro and function as a tissue-specific feature
in disease (10,24,25).
A number of disease-associated DNA methylation variations have been
identified in a diverse range of tissues, including the peripheral
blood (8,10,20,26).
T2D is a major metabolic disease, which
physiological and pathological states may be regulated by
epigenetic modifications that control gene expression (10). Hundreds of differential DNA
methylation CpGs of genes promoters have been identified in genomic
DNA isolated from pancreatic islets using Infinium methylation
assay technology (1). These
disease-specific methylation signatures also exist in human
accessible peripheral tissues (8).
A prospective study found that significant hypomethylation of young
individuals was associated with a higher risk of developing T2D in
later life (8). In the present
study, the DNA promoter methylation levels of three T2D candidate
genes (CALM2, CRY2 and CAMK1D) in the peripheral blood were
investigated, providing further information regarding DNA promoter
methylation regulation in T2D.
Low levels of DNA promoter methylation were
identified in the three genes in the healthy subjects and patients
with T2D (Table I). The majority
of the methylation correlation coefficients of the CpGs within
these genes were also not significant (Fig. 1). Two reasons may explain these
observations. In epigenetics, not all candidate genes are directly
regulated by DNA methylation, with other epigenetic factors,
including non-coding RNA and histone variants, also involved in the
regulation of gene expression (20,23).
Since T2D is a polygenic disease, the low DNA promoter methylation
levels of CALM2, CRY2 and CAMK1D genes may indicate that DNA
promoter methylation has no direct effect on gene function. In
addition, the methylation status was only investigated in the
peripheral blood, and DNA promoter methylation modification has
been demonstrated to exhibit a tissue-specific methylation pattern
(25). The insulin-2 gene, an
additional candidate gene for diabetes, was found to be
unmethylated in β cells, but methylated in other tissues (25). The identification of DNA
methylation profiling in pancreatic islets revealed that
differences in the methylation status of certain candidate genes
between T2D and non-diabetic patients were not present in blood
cells (1). The two aforementioned
reasons may explain the low DNA methylation levels of CALM2, CRY2
and CAMK1D observed in the present study.
In conclusion, low methylation levels of CALM2, CRY2
and CAMK1D were observed in the peripheral blood of the healthy
controls and T2D patients. These observations indicate that DNA
methylation may not be the primary regulatory mechanism of CALM2,
CRY2 and CAMK1D in T2D, and these methylation loci may not be
regarded as biomarkers for T2D. The present study provides further
information with regard to the epigenetic mechanisms and may
provide reference value in genetic-based pharmacological
development for future T2D treatments.
Acknowledgements
The study was supported by grants from the National
Natural Science Foundation of China (nos. 31100919 and 81371469),
the Natural Science Foundation of Zhejiang Province (no.
LR13H020003) and the K.C. Wong Magna Fund in Ningbo University,
Ningbo Social Development Research Projects (no. 2012C50032).
Abbreviations:
|
T2D
|
type 2 diabetes
|
|
CAMK1D
|
calcium/calmodulin-dependent protein
kinase 1D
|
|
CRY2
|
cryptochrome 2
|
|
CALM2
|
calmodulin 2
|
|
CI
|
confidence interval
|
|
ALT
|
alanine aminotransferase
|
|
SNP
|
single nucleotide polymorphism
|
|
PCR
|
polymerase chain reaction
|
References
|
1
|
Volkmar M, Dedeurwaerder S, Cunha DA, et
al: DNA methylation profiling identifies epigenetic dysregulation
in pancreatic islets from type 2 diabetic patients. EMBO J.
31:1405–1426. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Omori S, Tanaka Y, Takahashi A, et al:
Association of CDKAL1, IGF2BP2, CDKN2A/B, HHEX, SLC30A8, and KCNJ11
with susceptibility to type 2 diabetes in a Japanese population.
Diabetes. 57:791–795. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gu T, Horová E, Möllsten A, et al: IGF2BP2
and IGF2 genetic effects in diabetes and diabetic nephropathy. J
Diabetes Complications. 26:393–398. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kwak SH, Kim SH, Cho YM, et al: A
genome-wide association study of gestational diabetes mellitus in
Korean women. Diabetes. 61:531–541. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Grarup N, Rose CS, Andersson EA, et al:
Studies of association of variants near the HHEX, CDKN2A/B, and
IGF2BP2 genes with type 2 diabetes and impaired insulin release in
10,705 Danish subjects: validation and extension of genome-wide
association studies. Diabetes. 56:3105–3111. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jia H, Yu L, Jiang Z and Ji Q: Association
between IGF2BP2 rs4402960 polymorphism and risk of type 2 diabetes
mellitus: a meta-analysis. Arch Med Res. 42:361–367. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu J, Wu J, Zhou Y, et al: Quantitative
assessment of the variation in IGF2BP2 gene and type 2 diabetes
risk. Acta Diabetol. 49(Suppl 1): S87–S97. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Toperoff G, Aran D, Kark JD, et al:
Genome-wide survey reveals predisposing diabetes type 2-related DNA
methylation variations in human peripheral blood. Hum Mol Genet.
21:371–383. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fraga MF, Ballestar E, Paz MF, et al:
Epigenetic differences arise during the lifetime of monozygotic
twins. Proc Natl Acad Sci USA. 102:10604–10609. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Barres R and Zierath JR: DNA methylation
in metabolic disorders. Am J Clin Nutr. 93:897S–900S. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Barrès R, Osler ME, Yan J, et al: Non-CpG
methylation of the PGC-1alpha promoter through DNMT3B controls
mitochondrial density. Cell Metab. 10:189–198. 2009.PubMed/NCBI
|
|
12
|
Verploegen S, Ulfman L, van Deutekom HW,
et al: Characterization of the role of CaMKI-like kinase (CKLiK) in
human granulocyte function. Blood. 106:1076–1083. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shu XO, Long J, Cai Q, et al:
Identification of new genetic risk variants for type 2 diabetes.
PLoS Genet. 6:e10011272010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Imamura M, Iwata M, Maegawa H, et al:
Genetic variants at CDC123/CAMK1D and SPRY2 are associated with
susceptibility to type 2 diabetes in the Japanese population.
Diabetologia. 54:3071–3077. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schleinitz D, Tönjes A, Böttcher Y, et al:
Lack of significant effects of the type 2 diabetes susceptibility
loci JAZF1, CDC123/CAMK1D, NOTCH2, ADAMTS9, THADA, and TSPAN8/LGR5
on diabetes and quantitative metabolic traits. Horm Metab Res.
42:14–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Murea M, Lu L, Ma L, et al: Genome-wide
association scan for survival on dialysis in African-Americans with
type 2 diabetes. Am J Nephrol. 33:502–509. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dupuis J, Langenberg C, Prokopenko I, et
al: New genetic loci implicated in fasting glucose homeostasis and
their impact on type 2 diabetes risk. Nat Genet. 42:105–116. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ribel-Madsen R, Fraga MF, Jacobsen S, et
al: Genome-wide analysis of DNA methylation differences in muscle
and fat from monozygotic twins discordant for type 2 diabetes. PLoS
One. 7:e513022012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Alberti KG and Zimmet PZ: Definition,
diagnosis and classification of diabetes mellitus and its
complications. Part 1: diagnosis and classification of diabetes
mellitus provisional report of a WHO consultation. Diabet Med.
15:539–553. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
García-Giménez JL, Sanchis-Gomar F, Lippi
G, et al: Epigenetic biomarkers: A new perspective in laboratory
diagnostics. Clin Chim Acta. 413:1576–1582. 2012.PubMed/NCBI
|
|
21
|
Zampetaki A, Kiechl S, Drozdov I, et al:
Plasma microRNA profiling reveals loss of endothelial miR-126 and
other microRNAs in type 2 diabetes. Circ Res. 107:810–817. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao C, Dong J, Jiang T, et al: Early
second-trimester serum miRNA profiling predicts gestational
diabetes mellitus. PLoS One. 6:e239252011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dehwah MA, Xu A and Huang Q: MicroRNAs and
type 2 diabetes/obesity. J Genet Genomics. 39:11–18. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Poy MN, Eliasson L, Krutzfeldt J, et al: A
pancreatic islet-specific microRNA regulates insulin secretion.
Nature. 432:226–230. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Husseiny MI, Kuroda A, Kaye AN, Nair I,
Kandeel F and Ferreri K: Development of a quantitative
methylation-specific polymerase chain reaction method for
monitoring beta cell death in type 1 diabetes. PLoS One.
7:e479422012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kong L, Zhu J, Han W, et al: Significance
of serum microRNAs in pre-diabetes and newly diagnosed type 2
diabetes: a clinical study. Acta Diabetol. 48:61–69. 2011.
View Article : Google Scholar : PubMed/NCBI
|