Introduction
Sepsis is a complex clinical syndrome that is caused
by a harmful host response to infection. Numerous advances have
been made in antibiotic therapy and supportive care; however,
sepsis remains a major cause of mortality in intensive care units.
The lung is the most vulnerable organ during sepsis (1). During the development of sepsis,
bacterial components, such as lipopolysaccharide (LPS), may
activate an inflammatory cascade, which results in the release of
inflammatory mediators. The overproduction of inflammatory
mediators induces endothelial and epithelial injury, vascular
leakage, edema and vasodilatation, subsequently causing the
development of acute lung injury (ALI) and acute respiratory
distress syndrome (ARDS) (2,3).
Previous studies have shown that inflammatory mediators have a key
function in the pathogenesis of ALI/ARDS. ALI is characterized by a
local inflammatory response, and Toll-like receptor 4 (TLR4) has an
important function in the activation of innate immunity via
recognizing LPS (4,5). Based on the key role of inflammatory
mediators in the pathogenesis of ALI, recent treatments have been
aimed at modulating TLR4 signaling and alleviating nonspecific
inflammatory reactions that may result in potential therapeutic
advantages for ALI.
As an essential receptor for LPS signaling, TLR4 may
trigger the activation of an extracellular signaling pathway and
result in the upregulation of inflammatory mediators (1,6). A
previous study demonstrated that therapeutic antagonism of TLR4
signaling protects against ALI (7). To neutralize LPS signaling, the
specific TLR4 monoclonal antibody (mAb) was used in the present
study to evaluate the effect on an experimental model of ALI. It
was hypothesized that suppressing TLR4-associated production of
inflammatory mediators by pretreatment with TLR4 mAb may be a
promising therapeutic strategy for the treatment of ALI.
Materials and methods
Animals
A total of 45 male BALB/c mice, weighing between 18
and 20 g, were obtained from the Guangdong Province Medical
Experiments Animal Center. Mice were provided with free access to
water and standard rodent chow, and were housed in pathogen-free
cages. The study was performed in accordance with the
recommendations in the Guide for the Care and Use of Laboratory
Animals of the National Institutes of Health. The animal use
protocol was reviewed and approved by the Institutional Animal Care
and Use Committee of Sun Yat-sen Memorial Hospital of the
University of Sun Yat-sen (Guangzhou, China).
Experimental protocol
Mice were divided into three groups: Control (group
C), sepsis (group S) and pretreatment (group P) groups. Mice in the
P and S groups were intraperitoneally injected with 10 mg/kg LPS
(Sigma-Aldrich, St. Louis, MO, USA) to establish an ALI model
(8). Group P mice were also
intraperitoneally injected with TLR4 mAb (5 μg/g; GeneTex, Irvine,
CA, USA) 1 h prior to the injection of LPS. The mRNA expression
levels of TLR4 in the lung tissue, as well as the serum expression
levels of tumor necrosis factor (TNF)-α and interleukin (IL)-6, the
water content of the lung and the pathomorphological changes in the
lung, were detected after 6, 12 and 24 h.
Analysis of inflammatory mediators in the
serum
Mice were sacrificed via an intraperitoneal
injection of 120 mg/kg pentobarbital, and blood samples were
collected from the right atrium. The expression levels of TNF-α and
IL-6 in the serum were then determined using an enzyme-linked
immunosorbent assay (ELISA; Biolegend, San Diego, CA, USA), in
accordance with the manufacturer’s instructions.
Lung histology
Following euthanasia, the lungs were excised from
the mice by opening the chest via median sternotomy. The right
inferior lobe was removed and fixed in 10% buffered formalin for 24
h. Hematoxylin and eosin-stained sections were prepared using
standard techniques as described by Szaka et al (9). The degree of microscopic injury was
scored based on the following variables: Hemorrhage, edema,
exudation, necrosis, congestion, neutrophil infiltration and
atelectasis. The severity of injury was judged based on the
following criteria (10): No
injury, 0; injury to 25% of the field, 1; injury to 50% of the
field, 2; injury to 75% of the field, 3; and diffuse injury, 4. The
ultimate score was obtained by adding the aforementioned scores. A
pathologist, who was blinded to the experimental protocol, provided
a score for each variable based on the severity of injury (11,12).
Water content of lung
Following euthanasia, the right middle lobe was
excised from each mouse. The wet weight of the lung was measured
using an electronic scale and then desiccated in an oven at 85°C
for 48 h to determine the dry weight. The water content was
obtained using the following equation: Water content (%) = (wet
weight - dry weight)/wet weight × 100%.
Expression of TLR4 mRNA in the lung
tissue
The mRNA expression levels of TLR4 were analyzed by
quantitative polymerase chain reaction. Total RNA was isolated from
the lung tissue using TRIzol reagent (Invitrogen Life Technologies,
Carlsbad, CA, USA) and subsequently reverse-transcribed into cDNA
using Superscript II RNase H-Reverse Transcriptase (Invitrogen Life
Technologies). The sequence-specific primers used were as follows:
β-actin (sense), 5′-TACAGCTTCACCACCACAGC-3′ and antisense,
5′-AAGGAAGGCTGGAAGAGAGC-3′; TLR4 sense, 5′-TGCAATGTGAGCATTGATGA-3′
and antisense, 5′-TGACACCATTGAAGCTGAGG-3′.
Statistical analysis
All data are presented as the mean ± standard
deviation. GraphPad Prism 4.0 software (GraphPad Software, Inc.,
San Diego, CA, USA) was used for statistical analysis. Data were
analyzed using one-way analysis of variance (ANOVA) by comparing
the inter-group results and via two-way ANOVA by comparing the
intra-group results. P<0.05 was considered to indicate a
statistically significant difference.
Results
Water content in the lung tissue
As shown in Table
I, the changes in the water content in the lungs of the mice in
groups C, S and P were not statistically different at 6 h
(P>0.05). However, when the LPS treatment time was increased,
the water content in the lung tissue increased by ~13 and 16% at 12
and 24 h, respectively, as compared with group C. This result is in
accordance with the changes observed in ALI. Pretreatment with TLR4
mAb prior to treatment with LPS was found to correct the
LPS-induced increase in the water content of the lung tissue
(P<0.05).
 | Table IWater content in the lung tissue of
each group. |
Table I
Water content in the lung tissue of
each group.
| Parameter | 6 h | 12 h | 24 h |
|---|
| Group C (%) | 72.47±2.89 | 71.32±4.13 | 71.20±3.11 |
| Group S (%) | 77.63±4.13 | 84.29±4.69a,c | 87.23±5.12a,c,d |
| Group P (%) | 76.65±5.32 | 78.35±3.61a,b | 83.32±4.87a,b,c,d |
| F-value | 2.09 | 13.32 | 17.55 |
Expression of TNF-α in the serum
To evaluate the effect of TLR4 mAb on an LPS-induced
inflammatory mediator, the expression of TNF-α in the serum was
measured using ELISA. As shown in Table II, the expression of TNF-α in
group S significantly increased by 230 pg/ml maximally at 6 h
(P<0.05) when compared with group C. Compared with group S,
pretreatment with TLR4 mAb prior to LPS treatment reduced the
expression of TNF-α induced by LPS maximally by 100 pg/ml at 6 h
(P<0.05).
| Table IISerum expression levels of TNF-α in
each group. |
Table II
Serum expression levels of TNF-α in
each group.
| Parameter | 6 h | 12 h | 24 h |
|---|
| Group C (pg/ml) | 29.45±5.25 | 33.13±3.88 | 34.08±4.12 |
| Group S (pg/ml) | 259.12±19.81a | 186.21±13.75a,c | 115.89±17.91a,c,d |
| Group P (pg/ml) | 156.85±14.11a,b | 112.43±14.33a,b,c | 81.45±10.58a,b,c,d |
| F-value | 324.96 | 214.53 | 56.21 |
Expression of IL-6 in the serum
Changes in the serum expression levels of IL-6 were
also investigated, and the results were similar to those observed
with TNF-α. As shown in Table
III, the expression levels of IL-6 in group S significantly
increased by 310 pg/ml maximally at 24 h (P<0.05) when compared
with group C, and pretreatment with TLR4 mAb also effectively
inhibited the increase in TNF-α expression induced by LPS
(P<0.05). These results indicated that pretreatment with TLR4
mAb may correct the expression of LPS-induced inflammatory
mediators.
| Table IIISerum expression levels of IL-6 in
each group. |
Table III
Serum expression levels of IL-6 in
each group.
| Parameter | 6 h | 12 h | 24 h |
|---|
| Group C
(pg/ml) | 84.83±5.74 | 86.52±7.37 | 84.96±10.06 |
| Group S
(pg/ml) |
235.75±32.26a |
300.64±13.65a,c |
394.58±13.55a,c,d |
| Group P
(pg/ml) |
157.93±18.41a,b |
204.58±15.40a,b,c |
308.59±14.11a,b,c,d |
| F-value | 60.45 | 360.78 | 791.18 |
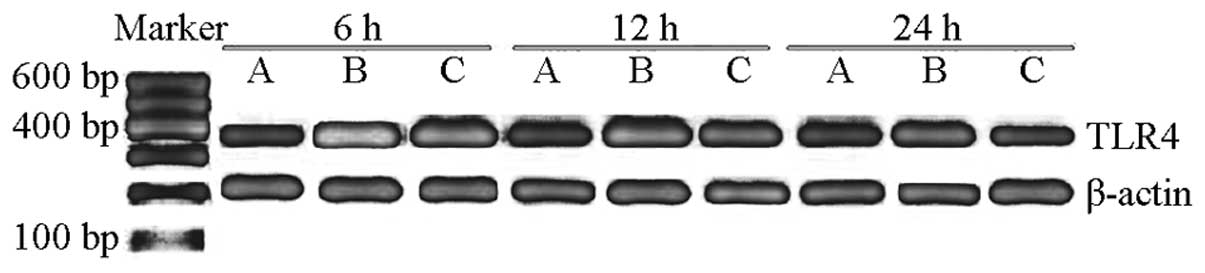
Expression of TLR4 mRNA in the lung
tissue
The mRNA expression level of TLR4 in group C was
0.304±0.03. The mRNA expression level of TLR4 in group S
significantly increased compared with that in group C (P<0.05),
and the peak value was observed at 6 h (1.697±0.090), which then
gradually decreased after 12 and 24 h (1.280±0.083 and 0.913±0.093,
respectively). However, the expression levels of TLR4 mRNA at 6, 12
and 24 h in group P were 1.201±0.053, 0.921±0.049 and 0.404±0.051,
respectively, thus, were significantly decreased compared with
group S (P<0.05; Table IV,
Fig. 1). These observations
indicated that TLR4 mAb may decrease the mRNA expression of TLR4 in
the lung tissue of sepsis mice.
| Table IVmRNA expression levels of
TLR4/β-actin in each group. |
Table IV
mRNA expression levels of
TLR4/β-actin in each group.
| Parameter | 6 h | 12 h | 24 h |
|---|
| Group C (%) | 0.304±0.031 | 0.315±0.022 | 0.309±0.025 |
| Group S (%) | 1.697±0.090a | 1.280±0.083a,c | 0.913±0.093a,c,d |
| Group P (%) | 1.201±0.053a,b | 0.921±0.049a,b,c | 0.404±0.051a,b,c,d |
| F-value | 70.21 | 58.35 | 36.48 |
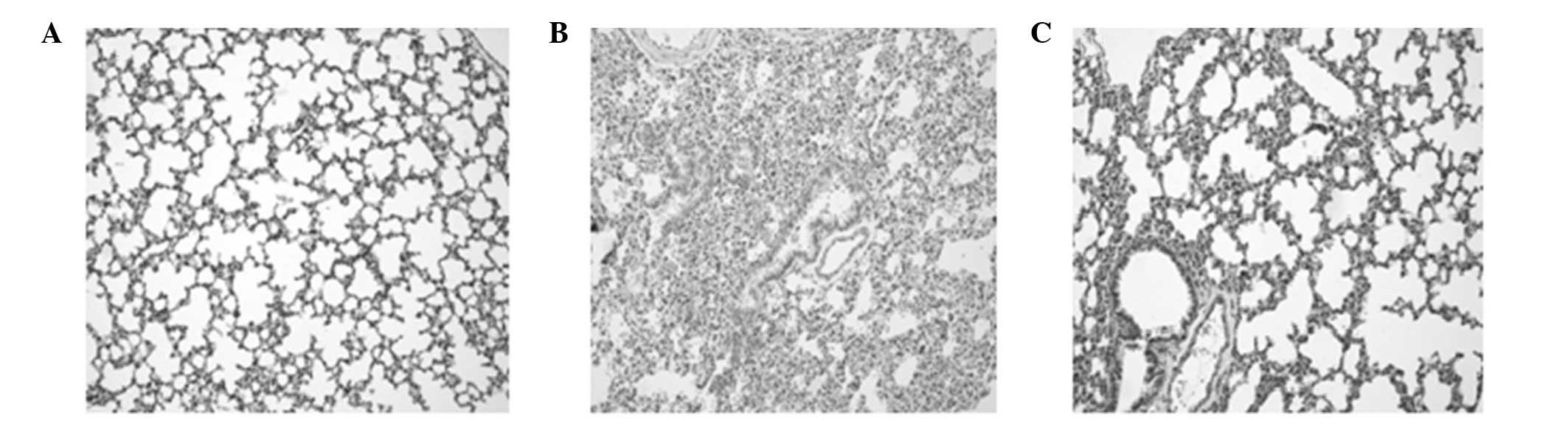
Pathological examination of the lung
tissue using light microscopy
The pulmonary organizational structure of group C
was normal. However, in group S at 6 and 12 h, the dilation of the
alveolus was not uniform since a number of the alveoli had
collapsed. Incrassation of the alveoli septum, neutrophil
infiltration and enlargement and congestion of the capillaries were
also observed. The extent of the pathological changes was
aggravated with increasing duration. In group S at 24 h, in
addition to the aforementioned pathological changes, airway trauma,
lung edema and local lung hemorrhage were observed. The
pathological changes in group P were significantly improved
compared with those in group S (Fig.
2).
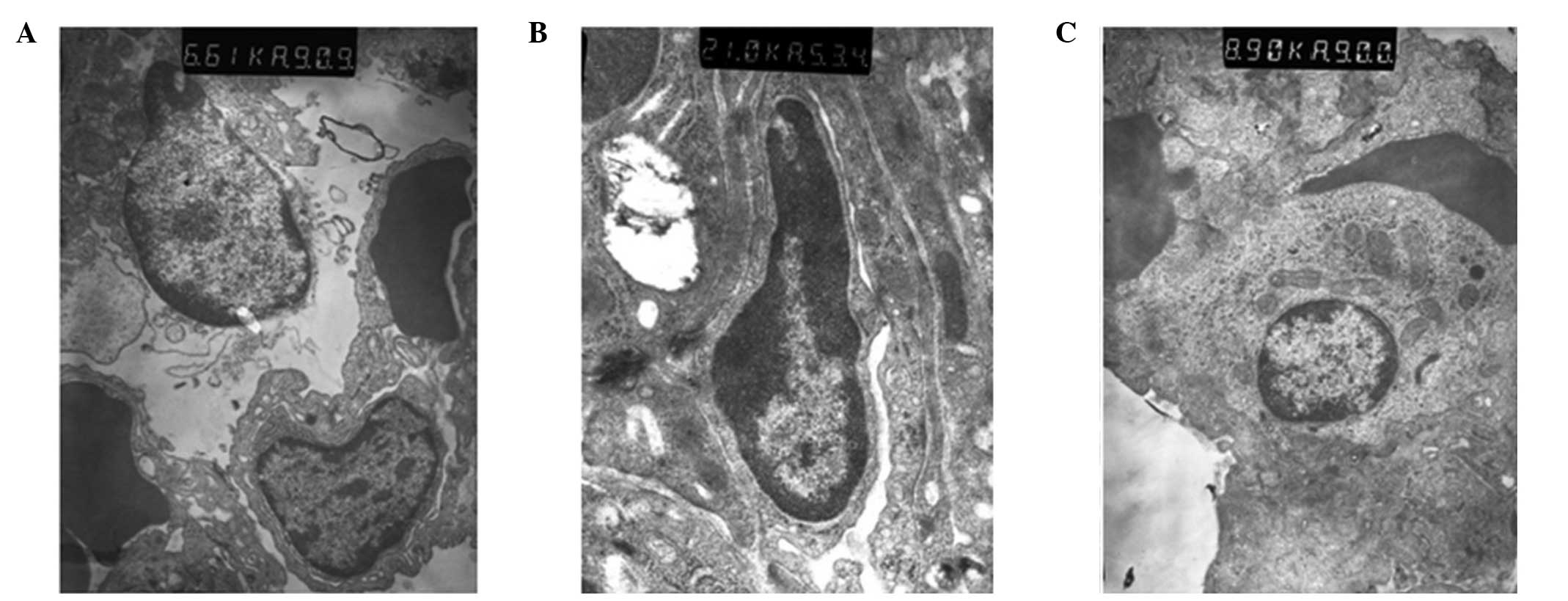
Ultrastructural changes of the lung
tissue observed using electron microscopy
In group C, the structure of the lung tissue and
alveolar were normal and clear, and edema and inflammatory changes
were observed. However, ultrastructural injury in the lung tissue
was observed in group S. In addition, alveolar epithelial cell
vacuolization and organelle swelling, dissolution and necrosis were
observed. The nuclei of the cells were twisted, and sections of the
chromatin were dissolved and had migrated to the edge of the cells.
Alveolar wall thickening and capillary expansion, as well as
pulmonary microvascular basement membrane thickening, were also
observed. Over time, rupture with widened alveolar septa and
pulmonary interstitial edema were observed, along with the
infiltration of the alveolar space by numerous red blood cells and
neutrophil granulocytes. The ultrastructural injury in group P was
milder compared with group S, in which pulmonary interstitial edema
was not observed and cell infiltration was reduced (Fig. 3).
Discussion
TLRs were identified in the late 1990′s as primary
sensors of microbial infection, and their identification led to
significant advances (13),
including that TLRs were broadly involved in transducing sensory
information in bacteria and TLR4 activated NF-κB triggers the
production of several inflammatory cytokines. Currently, 12 members
of the TLR family have been identified in mammalian cells (14). Among these, TLR4 detects the
presence of gram-negative bacteria through the recognition of the
lipid A moiety of LPS (15,16).
TLR4 is a type I integral membrane glycoprotein with a cytoplasmic
signaling domain that is homologous to the signaling domain of the
IL-1 receptor (IL-1R), known as the Toll/IL-1R homology (TIR)
domain (17). Following ligand
binding, TLR4 recruits the TIR-domain-containing adaptor molecules
to the TIR domain. Myeloid differentiation factor 88 (MyD88) is an
important adaptor molecule. Upon stimulation, MyD88 associates with
the cytoplasmic domain of TLR4 and subsequently recruits
IL-1R-associated kinase 1 (IRAK-1) and IRAK-4 (18–20),
which mediate the activation of the transcriptional factor, nuclear
factor (NF)-κB, leading to the induction of inflammatory mediators.
The initiation of the innate immune response through TLR4 triggers
an inflammatory cascade that is the primary cause of harmful
conditions, including ALI. Therefore, to suppress the inflammatory
cascade effectively, we hypothesized that the inhibition of a point
upstream of the TLR4 signaling pathway may be a better approach
than inhibiting each mediator. Abraham (19) used a TNF-α mAb to treat septic
shock, while Fisher (20) used an
IL-1R antagonist in the treatment of patients with sepsis syndrome;
however, the results were not satisfactory.
Previous studies have shown that the therapeutic
antagonism of TLR4 signaling provides protection against ALI.
Recently, Shirey et al (7)
found that Eritoran (E5564), an extremely potent TLR4 antagonist,
exhibits a highly protective effect when administered
therapeutically to mice with influenza-induced ALI (7). In addition, it has previously been
found that TLR4 mAb may function as a TLR4 antagonist and reduce
the expression of IL-1β in LPS-stimulated decidual cells (21,22).
Furthermore, a previous study demonstrated that TLR4 mAb reduces
LPS-induced cytokine production by inhibiting the NF-κB pathway in
cells (23). ALI is characterized
by a local inflammatory response, and the potential efficacy of
TLR4 mAb to treat ALI is based on the hypothesis that TLR4 mAb
suppresses the TLR4-associated inflammatory cascade of ALI. In the
present study, TLR4 mAb was shown to exhibit a protective effect on
LPS-induced ALI in mice. The expression levels of TNF-α and IL-6
decreased, whilst lung histology and edema were markedly improved.
However, the underlying mechanisms require further
investigation.
In the present study, significantly increased serum
levels of TNF-α and IL-6 were observed in group S when compared
with those in group C, indicating that TNF-α and IL-6 have an
important role in LPS-induced ALI. TNF-α is derived from activated
macrophages and functions via specific cell membrane-bound
receptors. Injecting TNF-α into experimental animals causes a
syndrome similar to septic shock (1,24,25).
TNF-α is released during the first 30–90 min following exposure to
LPS, which in turn activates a secondary level of the inflammatory
cascade, including cytokines, lipid mediators and reactive oxygen
species (1). IL-6 is produced by a
wide range of cells, including macrophages and endothelial cells,
in response to stimulation by factors, such as endotoxins and TNF-α
(26–28). IL-6 is an important factor that
amplifies the inflammatory reaction and stimulates the synthesis of
acute phase protein (29,30) Circulating levels of IL-6 are
excellent predictors of the severity of ALI/ARDS of various
etiologies (31). In the present
study, significantly decreased levels of TNF-α and IL-6 were
observed in group P when compared with those in group S. The
inhibition of TLR4 signaling by TLR4 mAb resulted in the
suppression of TNF-α and IL-6, which resulted in low-level lung
edema and injury. TLR4 signaling is a critical activator of immune
defense during infection, and the effective termination of TLR
signaling is essential to prevent detrimental systemic effects,
including septic shock (32).
Therefore, the signaling mechanism of TLR4 mAb, the regulation of
TLR4 expression on the cell surface and the transcriptional
induction of negative regulators, including IL-1R-associated kinase
and suppressor of cytokine signaling 1, requires further
investigation (33).
In conclusion, TLR4 plays a critical role in LPS
induced-ALI. TLR4 mAb reduces the secretion of inflammatory factors
and attenuates the degree of pulmonary edema, thus, exhibits
protective effects against LPS-induced ALI.
Acknowledgements
This study was supported by grants from the Natural
Science Foundation of Guangdong Province (No S2013010014805) and
Educational Commission of Guangdong Province (No.
2012B091100456).
References
|
1
|
Cohen J: The immunopathogenesis of sepsis.
Nature. 420:885–891. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ware LB and Matthay MA: The acute
respiratory distress syndrome. N Engl J Med. 342:1334–1349. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Densmore JC, Signorino PR, Ou J, et al:
Endothelium-derived microparticles induce endothelial dysfunction
and acute lung injury. Shock. 26:464–471. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cribbs SK, Matthay MA and Martin GS: Stem
cells in sepsis and acute lung injury. Crit Care Med. 38:2379–2385.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Imai Y, Kuba K, Neely GG, et al:
Identification of oxidative stress and Toll-like receptor 4
signaling as a key pathway of acute lung injury. Cell. 133:235–249.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu Y, Jagannath C, Liu XD, Sharafkhaneh A,
Kolodziejska KE and Eissa NT: Toll-like receptor 4 is a sensor for
autophagy associated with innate immunity. Immunity. 27:135–144.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shirey KA, Lai W, Scott AJ, et al: The
TLR4 antagonist Eritoran protects mice from lethal influenza
infection. Nature. 497:498–502. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tong Q, Zheng L, Kang Q, Dodd-O J, Langer
J, Li B, Wang D and Li D: Upregulation of hypoxia-induced mitogenic
factor in bacterial lipopolysaccharide-induced acute lung injury.
FEBS Lett. 580:2207–2215. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Szarka RJ, Wang N, Gordon L, Nation PN and
Smith RH: A murine model of pulmonary damage induced by
lipopolysaccharide via intranasal instillation. J Immunol Methods.
202:49–57. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mrozek JD, Smith KM, Bing DR, Meyers PA,
Simonton SC, Connett JE and Mammel MC: Exogenous surfactant and
partial liquid ventilation: physiologic and pathologic effects. Am
J Respir Crit Care Med. 156:1058–1065. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rotta AT and Steinhorn DM: Partial liquid
ventilation reduces pulmonary neutrophil accumulation in an
experimental model of systemic endotoxemia and acute lung injury.
Crit Care Med. 26:1707–1715. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nahum A, Hoyt J, Schmitz L, Moody J,
Shapiro R and Marini JJ: Effect of mechanical ventilation strategy
on dissemination of intratracheally instilled Escherichia
coli in dogs. Crit Care Med. 25:1733–1743. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rock FL, Hardiman G, Timans JC, Kastelein
RA and Bazan JF: A family of human receptors structurally related
to Drosophila Toll. Proc Natl Acad Sci USA. 95:588–593.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Akira S, Uematsu S and Takeuchi O:
Pathogen recognition and innate immunity. Cell. 124:783–801. 2006.
View Article : Google Scholar
|
|
15
|
Kumar H, Kawai T and Akira S: Pathogen
recognition by the innate immune system. Int Rev Immunol. 30:16–34.
2011. View Article : Google Scholar
|
|
16
|
Huang S, Miao R, Zhou Z, et al: MCPIP1
negatively regulates Toll-like receptor 4 signaling and protects
mice from LPS-induced septic shock. Cell Signal. 25:1228–1234.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jenkins KA and Mansell A: TIR-containing
adaptors in Toll-like receptor signalling. Cytokine. 49:237–244.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
López-Bojórquez LN, Dehesa AZ and
Reyes-Terán G: Molecular mechanisms involved in the pathogenesis of
septic shock. Arch Med Res. 35:465–479. 2004.
|
|
19
|
Abraham E, Anzueto A, Gutierrez G, et al:
Double-blind randomised controlled trial of monoclonal antibody to
human tumour necrosis factor in treatment of septic shock. NORASEPT
II Study Group Lancet. 351:929–933. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fisher CJ Jr, Dhainaut JF, Opal SM, et al:
Recombinant human interleukin 1 receptor antagonist in the
treatment of patients with sepsis syndrome. Results from a
randomized, double-blind, placebo-controlled trial Phase III
rhIL-1ra Sepsis Syndrome Study Group. JAMA. 271:1836–1843. 1994.
View Article : Google Scholar
|
|
21
|
O‘Neill LA and Bowie AG: The family of
five: TIR-domain-containing adaptors in Toll-like receptor
signalling. Nat Rev Immunol. 7:353–364. 2007.
|
|
22
|
Li Y, Zhong S and Yao R: Influence of LPS
and Toll-like receptor 4 antagonist on progesterone receptor,
interleukin-1β, and cyclooxygenase-2 in decidual cells. Zhong Nan
Da Xue Xue Bao Yi Xue Ban. 38:162–168. 2013.(In Chinese).
|
|
23
|
Tsukamoto H, Fukudome K, Takao S,
Tsuneyoshi N, Ihara H, Ikeda Y and Kimoto M: Multiple potential
regulatory sites of TLR4 activation induced by LPS as revealed by
novel inhibitory human TLR4 mAbs. Int Immunol. 24:495–506. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Norman JG, Fink GW and Franz MG: Acute
pancreatitis induces intrapancreatic tumor necrosis factor gene
expression. Arch Surg. 130:966–970. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Putensen C and Wrigge H:
Ventilator-associated systemic inflammation in acute lung injury.
Intensive Care Med. 26:1411–1413. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bhatia M, Brady M, Shokuhi S, Christmas S,
Neoptolemos JP and Slavin J: Inflammatory mediators in acute
pancreatitis. J Pathol. 190:117–125. 2000. View Article : Google Scholar
|
|
27
|
Bhatia M, Neoptolemos JP and Slavin J:
Inflammatory mediators as therapeutic targets in acute
pancreatitis. Curr Opin Investig Drugs. 2:496–501. 2001.PubMed/NCBI
|
|
28
|
Bhatia M: Novel therapeutic targets for
acute pancreatitis and associated multiple organ dysfunction
syndrome. Curr Drug Targets Inflamm Allergy. 1:343–351. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Geiger T, Andus T, Klapproth J, Hirano T,
Kishimoto T and Heinrich PC: Induction of rat acute-phase proteins
by interleukin 6 in vivo. Eur J Immunol. 18:717–721. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Castell JV, Gómez-Lechón MJ, David M, et
al: Interleukin-6 is the major regulator of acute phase protein
synthesis in adult human hepatocytes. FEBS Lett. 242:237–239. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Remick DG, Bolgos GR, Siddiqui J, Shin J
and Nemzek JA: Six at six: interleukin-6 measured 6 h after the
initiation of sepsis predicts mortality over 3 days. Shock.
17:463–467. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Szatmary Z: Molecular biology of Toll-like
receptors. Gen Physiol Biophys. 31:357–366. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liew FY, Xu D, Brint EK and O‘Neill LA:
Negative regulation of Toll-like receptor-mediated immune
responses. Nat Rev Immunol. 5:446–458. 2005. View Article : Google Scholar : PubMed/NCBI
|