Introduction
Amniotic fluid contains a number of living cells
that have undergone defluxion from the fetus and fetal membranes
(1). As the amniotic fluid cells
(AFCs) share the same genomic background as the fetus, they
represent an important prenatal diagnosis target (2–8).
Amniotic fluid has been used for diagnosing genetic disease
(8), cytogenetic analysis
(5,7) and comparative genomic hybridization
(CGH) assays (6). As the process
of amniocentesis causes trauma, a degree of maternal blood
interference is unavoidable. Since blood cells are unable to
proliferate or adhere in culture in the absence of stimulus
factors, it is necessary to optimize the culture to specifically
amplify fetal cells in order to minimize maternal interference,
collect metaphase cells and acquire sufficient genomic DNA.
Previous studies have focused on stem cells derived
from amniotic fluid. Initially, AFCs were identified as glial cells
and monocyte-derived macrophages (9). Furthermore, it was presumed that
mesenchymal stem cells were derived from the AFCs (10) on the basis of their cell
morphology, specific surface markers and their capacity for neural,
adipogenic and osteogenic differentiation. A study by De Coppi
et al maintained pluripotent-specific cells in pluripotent
stem cell culture medium. These cells were positive for CD markers,
expressed pluripotent stem cell markers and were able to
differentiate into three germ lines (11). Additional studies have demonstrated
that AFCs are easily reprogrammed into induced pluripotent stem
cells (iPSCs) (12–14). As a result of their ability to
self-renew and differentiate into functional somatic cells,
amniotic fluid-derived stem cells have been used as seed cells for
tissue engineering (11,15,16)
and iPSCs have been used to model genetic disease (13,17).
Due to the wide application of AFCs in prenatal
diagnosis and their potential use as stem cells, the amplification
and identification of the characteristics of AFC cultures is
important for clinical laboratory diagnosis and biological study.
In the present study, surface markers, chromosome Giemsa
(G)-banding, cell cycle distribution and the proliferation of AFCs
in an early- and late-passage were assessed.
Materials and methods
Cell culture
Three independent amniotic fluid samples from two
females at 20 weeks and one female at 21 weeks into the gestation
period, who took part in a clinical cytogenetic diagnosis, were
included in the study. The patients provided consent and the study
was approved by the Ethics Committee of The Third Affiliated
Hospital of Guangzhou Medical University (Guangzhou, China). A
total of 16 ml amniotic fluid was extracted from each patient by
amniocentesis for cytogenetic diagnosis. Each sample was separated
into two tubes of 8 ml each. Following centrifugation at 300 × g
for 5 min, the cell precipitate in each tube was collected for
culture in a 25 cm2 flask [passage (P)0; Gibco
AmnioMAX™-C100; Invitrogen Life Technologies, Carlsbad,
CA, USA]. The cells were passaged for nine days following
inoculation in 0.05% trypsin-EDTA (Invitrogen Life Technologies).
Subsequently, one tube was seeded into an additional 25
cm2 flask for G-banding analysis, while the other tube
was seeded into a 75 cm2 flask for further experiments.
The subcultured cells were passaged four times every 5–6 days, when
80–90% of cells were confluent. Cells from P1 and P6 were regarded
as early- and late-passages, respectively, and their analyses were
compared.
G-banding
Cultured AFCs were incubated in 25 mg/ml colchicine
for 4 h and harvested using 0.05% trypsin-EDTA. The cells were made
hypotonic using hypotonic medium [0.4% sodium citrate combined with
0.4% potassium chloride (1:1)], fixed in a fixation medium [acetic
acid combined with methanol (1:3)], dropped on precooled glass and
cultured at 60°C overnight. G-banded karyotyping was performed by
digesting the cells with trypsin, followed by staining with a
Giemsa stain. Images were captured and analyzed using an Ikaros
system (Carl Zeiss AG, Oberkochen, Germany).
Proliferation curve rendering
For each individual culture, 1×105 cells
were seeded into nine independent 9.6 cm2 wells to
analyze their proliferative potential. After 2 h allowing for
adherence, cells from three wells were trypsinized and counted at
24, 48 and 72 h. As there were no statistically significant
difference between each individual. The mean values and standard
deviations of each time were used to construct a proliferation
curve. This experimental protocol was performed for P1 and P6
cells.
Flow cytometry analysis
Adherent AFCs were separated by trypsin treatment
and fixed in 75% ethanol overnight at 4°C. The cells were filtered
through a 40 μm mesh and resuspended in fluorescence-activated cell
sorting (FACS) buffer [phosphate-buffered saline (PBS) containing
2% fetal bovine serum (FBS) and 0.1% sodium azide].
Directly-conjugated isotype control antibodies, IgG-fluorescein
isothiocyanate (FITC) and IgG-phycoerythrin (PE; BD Pharmingen, San
Diego, CA, USA), were used as controls to identify the background
cells. A total of ~5×105 cells were incubated at 4°C for
40 min with each of the following FITC- or PE-conjugated antibodies
(BD Pharmingen): CD133-PE, CD117-PE, CD34-PE, CD105-FITC,
CD106-FITC, CD29-PE, CD44-FITC, CD147-FITC and CD90-PE. The cells
were subsequently washed in FACS buffer. The antibody-labeled cells
were analyzed using a BD FACSCalibur instrument (BD Biosciences,
Franklin Lakes, NJ, USA), and data were analyzed using FlowJo
version 7.2.5 software (TreeStar, Inc., Ashland, OR, USA). The cell
count experiments for the flow cytometry assays were performed in
triplicate.
Cell cycle distribution
AFCs were harvested and fixed using the same method
as described previously for flow cytometry analysis. Cellular DNA
was stained with propidium iodide (PI) at 4°C for 30 min in a
staining solution composed of PBS with 50 μg/ml PI, 100 μg/ml RNase
A and 0.2% Triton X-100. The cells were counted using a BD
FACSCalibur instrument. ModFit software (BD Biosciences) was used
to conduct analysis.
Statistical analysis
For P1 and P6, nine independent cultures of each AFC
were performed. The nine cultures were divided into three groups,
three cultures were harvested at 24, three at 48 and three at 72 h.
Differences between the individual cultures were analyzed using a
t-test. In addition, the CD+ percentage and percentage
of AFCs in each cell cycle stage for the early- and late-passage
cultures were compared using the t-test. All statistical analyses
were performed by SPSS statistical software (version 19.0, IBM SPSS
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
AFCs exhibit different morphologies in
primary culture
Following seven days of primary culture, a number of
cells adhered to form cell colonies, whereas other cells remained
suspended and were removed when the medium was changed. The cell
colonies were divided into two types according to their morphology:
Endothelial and fibroblast-like. Endothelial colonies were
characteristically polygonal, large cells with a tight cell
connection and a legible colony edge. By contrast, the
fibroblast-like colonies comprised slender, spindle-shaped cells
that did not possess a tight connection and typically exhibited a
number of cells away from the colony edge (Fig. 1). After nine days of primary
culture, as the fibroblast cells grew more rapidly than the
endothelial cells, certain fibroblast-like colonies became
confluent with one other or with the endothelial colonies. At this
stage, the primary culture cells were trypsinized and termed as
P1.
Early-passage AFCs exhibit a mesenchymal
cell marker and a normal karyotype
Following the primary AFC subculture, the adherent
cells remained distinguishable as fibroblast or endothelial cells,
and certain small endothelial colonies were apparent. A number of
the P1 cells were harvested for karyotype G-banding, cell surface
marker or cell cycle distribution by flow cytometry analyses. The
remaining cells were cultured for cell proliferation analysis and
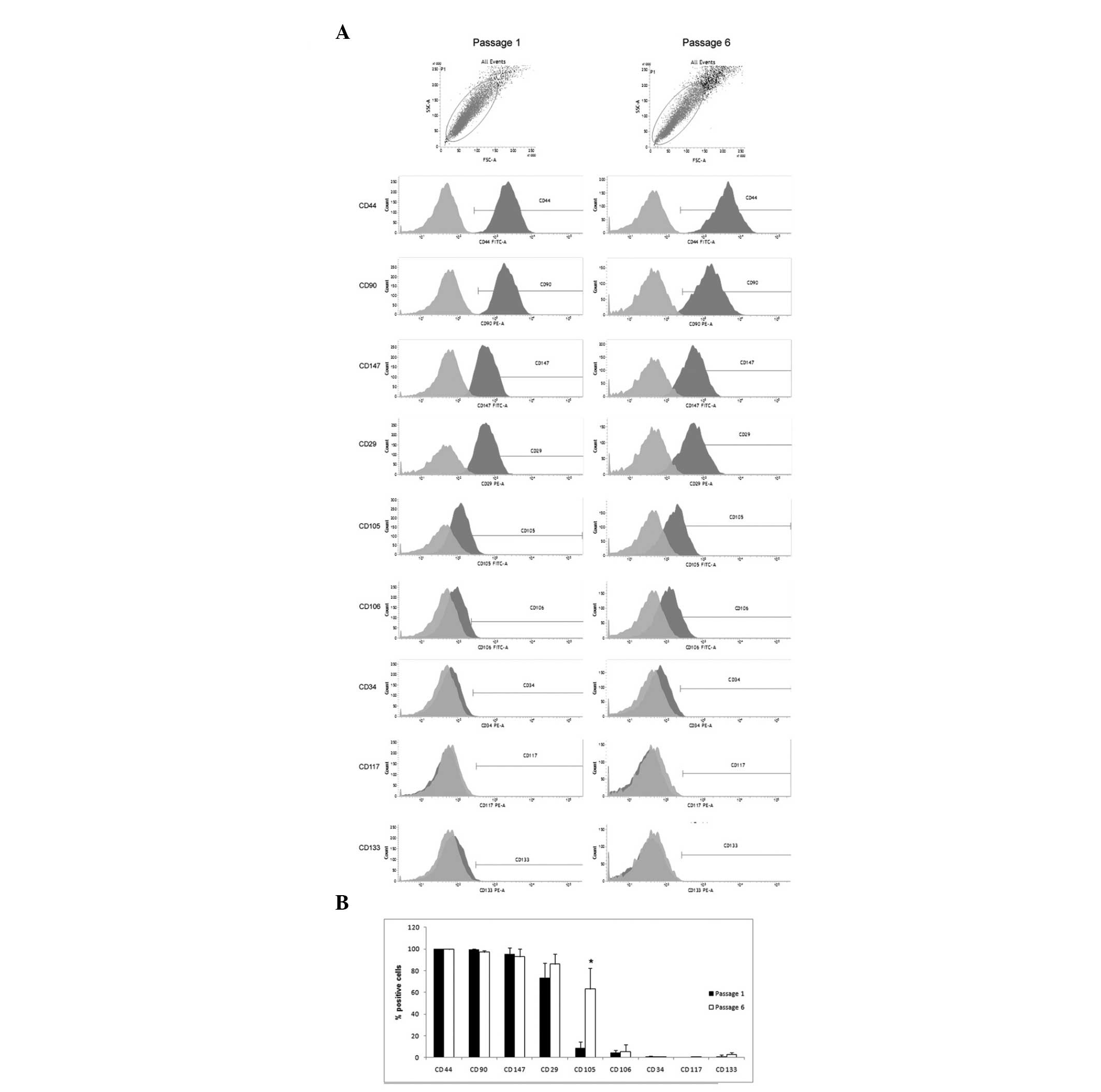
long-term culture. Although the percentage of CD+ cells
varied among the three independent cultures, the P1 cells were
almost all (>85%) positive for the mesenchymal cell markers,
CD44, CD90 and CD147, partially positive for CD29, CD105 and CD106,
but negative (<1.5%) for CD117, CD34 and CD133 (Figs. 2 and 3). G-banding analysis revealed that the
three AFC cultures exhibited normal karyotypes (Fig. 1) and were at the following
cell-cycle stages: G1 (79.27%), S (7.63%) and G2/M (13.1%; Fig. 4). The proliferative ability was
obtained by quantifying the number of cells in culture, and
statistically significant differences were not observed among the
AFC cultures following t-test analysis. The time taken for the
cells to double in quantity was 24 h (Fig. 1).
Late-passage AFCs retain a normal
karyotype, but cell surface markers change, cell cycle is blocked
in S-phase and proliferation ability is reduced
The late-passage of AFCs refers to cells that have
undergone six passages, and during the nearly 30 day culture there
was 30 times doubling. G-banding analysis revealed that the three
AFC groups maintained a normal karyotype (Fig. 1). With regard to morphology, the
tight endothelial colonies did not exist, and the cells exhibited a
homogeneous morphology. The late-passage AFCs were similar to the
early-passage AFCs in that they expressed almost all the same
mesenchymal cell markers, including CD44, CD90 and CD147, were
partially positive for CD29, CD105 and CD106, but negative for
CD117, CD34 and CD133. However, the percentage of CD105+
cells in the late-passage AFC cultures was significantly higher
compared with the early-passage cells (P<0.05; Fig. 2 and 3). The AFCs exhibited the following cell
cycle stage distributions: G1 (67.59%), S (20.49%) and G2/M
(11.91%). On average, the percentages of late-passage AFCs in the
G1 and G2/M phases were not significantly different when compared
with the early-passage AFCs; however, the percentage of cells in
the S-phase was significantly higher in the late-passage cells
compared with the early-passage cells (P<0.05; Fig. 4). The proliferative ability of the
AFCs exhibited statistically significant differences between the
passages when the doubling time was 24–48 h (Fig. 1).
Discussion
AFC cultures are not only required for traditional
prenatal diagnosis, but are also widely used for purifying fetal
samples in order to avoid interference from maternal material when
genetically diagnosing diseases or performing CGH assays (2,3,6–8).
Furthermore, a number of studies on stem cells and tissue
engineering have indicated that AFCs are suitable for acquiring
human genetic material for disease diagnosis (12,13)
and for use as seed cells in tissue engineering (11,15,16).
In the present study, three AFC samples from pregnant females in
their second trimester were included for a duration that was
suitable for prenatal diagnosis based on amniotic fluid (18–20).
Previously, primary AFCs were regarded as
colonial-morphology growth cells, while fetal urine was considered
a potential source of AFCs (18,19).
The morphology of the colonies was separated into two or three
divisions. The two divisions were F-type (identical to cultured
human dermal fibroblasts) and AF-type (amniotic fluid type),
characterized by the presence of type I collagen fibers (1), while the three division were
fibroblast-like (F), epithelioid (E) and clonable AFCs (20). In the current study, the colonies
in the primary culture were divided into endothelial or
fibroblast-like colonies; all of which originated from a fetal
source and were able to proliferate.
As the amount of primary AFC culture acquired was
not sufficient to perform all the subsequent analyses, the AFCs
were subcultured. In the subculture, few suspended cells were
discarded and the incubation time from primary culture to P1 was 48
h. The P1 cell type was considered to be similar to the primary
culture. A clinical cytogenetic diagnosis was performed on the P1
cells and the three independent cultures were demonstrated to
exhibited a normal karyotype. Flow cytometry analysis results
indicated that almost all the cells were positive for CD44, CD90
and CD147 and almost all were negative for CD34, CD117 and CD133. A
number of the AFC population was positive for CD29, CD105 and
CD106, with a variety of frequencies for each. The clinically
cultured AFCs were positive for mesenchymal cell markers and
negative for endothelial, hematopoietic and stem cells markers.
This observation may be due to the culture medium and state, which
is consistent with the results of a previous study on cell
phenotypes (21,22). Several studies have derived
hematopoietic stem cells from amniotic fluid and performed
screening using flow cytometry prior to establishing the primary
culture (11). These cells were
discarded in the primary culture in the current study. The AFCs
were able to proliferate well and doubled in abundance within 24 h;
on average, 13.1% of the culture was in the mitotic phase. The main
purpose for clinically culturing AFCs has been for cytogenetic
analysis (5,20). In the present study, the clinical
culture conditions were suitable for this purpose as there were a
sufficient number of cells in the G2/M phase.
The present subculture was based on cell growth,
where following six passages and almost 30 divisions, the AFCs
retained a normal karyotype. The identification experiment was
performed by long-term culture comparison. With regard to the cell
surface markers, only the percentage of CD105+ cells
significantly increased in the late-passage when compared with the
early-passage AFC cultures; the other markers were not
significantly changed. Considering that CD105 is a mesenchymal
marker, the long-term culture conditions are permissive for
mesenchymal cell growth or for mesenchymal progenitor cells to
differentiate into CD105+ cells. Furthermore, the
clinical culture system was similar to a previously published
amniotic fluid stem cell study system (23), where amniotic fluid stem cells were
derived to perform adipogenic, osteogenic, neurogenic and myogenic
differentiation. These cells also expressed mesenchymal and neural
markers, but not hematopoietic markers. In the present study, the
late-passage cells proliferated at a slower rate compared with the
early-passage cells, evident from the doubling time, which was
>24 h but <48 h, but the doubling time was 24 h for the
early-passage cells. There were a sufficient number of mitotically
dividing cells to conduct karyotype analysis, consistent with the
11.91% of cells in a mitotic stage. Cell cycle distribution
analysis indicated that the percentage of cells in the S-phase
significantly increased during the late-passage, whereas the
percentage at other stages were not significantly different. It was
hypothesized that the cells were proliferating slowly due to
S-phase arrest in the late-passage cells.
In conclusion, the present study characterized the
exchange of surface markers and cell cycle distribution in an AFC
culture. It demonstrated hat the number of CD105+ AFCs
increased following long-term culture and the AFC proliferation
decreased due to cell cycle arrest in the S-phase. As the AFC
culture was widely used in tissue engineering and clinical prenatal
diagnosis, our study would be useful in the specific mesenchymal
cell acquisition and prenatal diagnosis sample keeping.
Acknowledgements
The study was supported by grants from the National
Natural Science Foundation of China (no. 81202604), the new teacher
project of the Education Ministry (no. 20134423120005), the China
Postdoctoral Science Foundation (no. Q088), the Guangzhou City
Science and Technology Administration (no. 2011y-00038-1), the
Guangdong Province Health Department (no. B2012169), the Guangdong
Province Natural Science Foundation (no. S2013040012649 and
S2013010014781), the Guangdong Province Higher Education Foundation
(2013KJCX0149 and Yq2013135), the Guangzhou Medical University (no.
2011C41 and 2012C63), and the Guangdong Province Reproductive and
Genetic Key Laboratory Foundation (no. 2012Z06). In addition, the
study was supported by the Guangdong Provincial Key Laboratory of
Malignant Tumor Epigenetics and Gene Regulation (Sun Yat-sen
Memorial Hospital, Sun Yat-sen University, Guangzhou, China).
References
|
1
|
Priest RE, Marimuthu KM and Priest JH:
Origin of cells in human amniotic fluid cultures: ultrastructural
features. Lab Invest. 39:106–109. 1978.PubMed/NCBI
|
|
2
|
Macek M, Hurych J, Hyánek J, et al:
Cultivation of amniotic fluid cells and prenatal diagnosis. Cesk
Gynekol. 36:556–558. 1971.(In Czech).
|
|
3
|
Mulcahy MT and Jenkyn J: Prenatal
diagnosis. Results of cytogenetic analysis of amniotic fluid cell
cultures. Med J Aust. 1:979–982. 1973.PubMed/NCBI
|
|
4
|
Macek M, Suk V, Bresták M, Rezácová D,
Houstĕk J and Kotásek A: Cultivation of cells from amniotic fluid
for prenatal diagnosis. Cesk Pediatr. 29:87–89. 1974.(In
Czech).
|
|
5
|
Zolotukhina TV: Amniotic fluid cell
culture for the prenatal diagnosis of the fetal karyotype. Tsitol
Genet. 14:57–63. 1980.(In Russian).
|
|
6
|
Lapierre JM, Cacheux V, Collot N, et al:
Comparison of comparative genomic hybridization with conventional
karyotype and classical fluorescence in situ hybridization for
prenatal and postnatal diagnosis of unbalanced chromosome
abnormalities. Ann Genet. 41:133–140. 1998.
|
|
7
|
Guven MA, Ceylaner G and Coskun A: Volume
of sampled amniotic fluid and prenatal cytogenetic diagnosis. Int J
Gynaecol Obstet. 95:157–158. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rebello MT, Hackett G, Smith J, et al:
Extraction of DNA from amniotic fluid cells for the early prenatal
diagnosis of genetic disease. Prenat Diagn. 11:41–46. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Polgár K, Adány R, Abel G, Kappelmayer J,
Muszbek L and Papp Z: Characterization of rapidly adhering amniotic
fluid cells by combined immunofluorescence and phagocytosis assays.
Am J Hum Genet. 45:786–792. 1989.PubMed/NCBI
|
|
10
|
In ‘t Anker PS, Scherjon SA, Kleijburg-van
der Keur C, et al: Amniotic fluid as a novel source of mesenchymal
stem cells for therapeutic transplantation. Blood. 102:1548–1549.
2003.PubMed/NCBI
|
|
11
|
De Coppi P, Bartsch G Jr, Siddiqui MM, et
al: Isolation of amniotic stem cell lines with potential for
therapy. Nat Biotechnol. 25:100–106. 2007.PubMed/NCBI
|
|
12
|
Li C, Zhou J, Shi G, et al: Pluripotency
can be rapidly and efficiently induced in human amniotic
fluid-derived cells. Hum Mol Genet. 18:4340–4349. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fan Y, Luo Y, Chen X, Li Q and Sun X:
Generation of human β-thalassemia induced pluripotent stem cells
from amniotic fluid cells using a single excisable lentiviral stem
cell cassette. J Reprod Dev. 58:404–409. 2012.
|
|
14
|
Li Q, Fan Y, Sun X and Yu Y: Generation of
induced pluripotent stem cells from human amniotic fluid cells by
reprogramming with two factors in feeder-free conditions. J Reprod
Dev. 59:72–77. 2013.PubMed/NCBI
|
|
15
|
Mirabella T, Cilli M, Carlone S, Cancedda
R and Gentili C: Amniotic liquid derived stem cells as reservoir of
secreted angiogenic factors capable of stimulating
neo-arteriogenesis in an ischemic model. Biomaterials.
32:3689–3699. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mirabella T, Poggi A, Scaranari M, et al:
Recruitment of host’s progenitor cells to sites of human amniotic
fluid stem cells implantation. Biomaterials. 32:4218–4227.
2011.
|
|
17
|
Lu HE, Yang YC, Chen SM, et al: Modeling
neurogenesis impairment in Down syndrome with induced pluripotent
stem cells from trisomy 21 amniotic fluid cells. Exp Cell Res.
319:498–505. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hoehn H, Bryant EM, Karp LE and Martin GM:
Cultivated cells from diagnostic amniocentesis in second trimester
pregnancies. I Clonal morphology and growth potential. Pediatr Res.
8:746–754. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hoehn H, Bryant EM, Fantel AG and Martin
GM: Cultivated cells from diagnostic amniocentesis in second
trimester pregnancies. III The fetal urine as a potential source of
clonable cells. Humangenetik. 29:285–290. 1975.PubMed/NCBI
|
|
20
|
Hoehn H, Bryant EM, Karp LE and Martin GM:
Cultivated cells from diagnostic amniocentesis in second trimester
pregnancies. II Cytogenetic parameters as functions of clonal type
and preparative technique. Clin Genet. 7:29–36. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Davydova DA, Vorotelyak EA, Smirnova YA,
et al: Cell phenotypes in human amniotic fluid. Acta Naturae.
1:98–103. 2009.PubMed/NCBI
|
|
22
|
Cananzi M and De Coppi P: CD117(+)
amniotic fluid stem cells: state of the art and future
perspectives. Organogenesis. 8:77–88. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bossolasco P, Montemurro T, Cova L, et al:
Molecular and phenotypic characterization of human amniotic fluid
cells and their differentiation potential. Cell Res. 16:329–336.
2006. View Article : Google Scholar : PubMed/NCBI
|