Introduction
Langerhans cell histiocytosis (LCH), previously
known as histiocytosis X, is a proliferative disease of
histiocyte-like cells. The condition is a rare disorder with
several forms of clinical presentations, which include osteolysis,
ulcerations of the skin and soft tissues, and occasionally viscera
involvement (1). The majority of
cases present as uni- or multifocal bone lesions, while visceral
involvement is less common (2,3). The
most frequent sites of a visceral manifestation are the lungs,
followed by the skin and the lymph nodes (2–5). The
etiology of LCH is yet to be completely understood, although
certain forms of immunological dysfunctions have been implicated.
LCH can occur at any age; however, the peak incidence is observed
in individuals aged between 1 and 3 years (6). A diagnosis of LCH should be based on
the synthetic analysis of clinical presentations, as well as
features of imaging and histopathology. Prognosis is dependent on a
variety of factors, including the age of onset, the number of
organs involved, the degree to which normal function of the organs
is affected and the rate of disease progression (7,8). For the
majority of children with LCH, the disease is self-resolving.
However, for patients with multisystemic involvement, the most
common treatment method is steroids and chemotherapeutic agents.
The present study reports the case of an infant diagnosed with LCH
with multisystem involvement, including that of the bone, skin,
orbit, spleen and lungs.
Case report
Written informed consent was obtained from the
patient's mother. An 11-month-old Chinese boy was admitted to the
China-Japan Union Hospital of Jilin University (Changchun, China)
following presentations of recurrent seborrheic dermatitis all over
the skin, bilateral otorrhea and a mass on the scalp. The
seborrheic dermatitis deteriorated, and ulcers developed on the
skin of the bilateral groin. In addition, the patient had a high
fever of up to 103°F. A decreased willing to stand or cry was
observed when the patient was standing, along with exophthalmos of
both eyes, particularly in the left eye. Physical examination
revealed a soft mass, measuring 1.5 cm in diameter, in the right
parietal region. Furthermore, computed tomography (CT) scans
demonstrated a round-shaped osteolytic lesion of the right parietal
bone (Fig. 1) and erosion of the
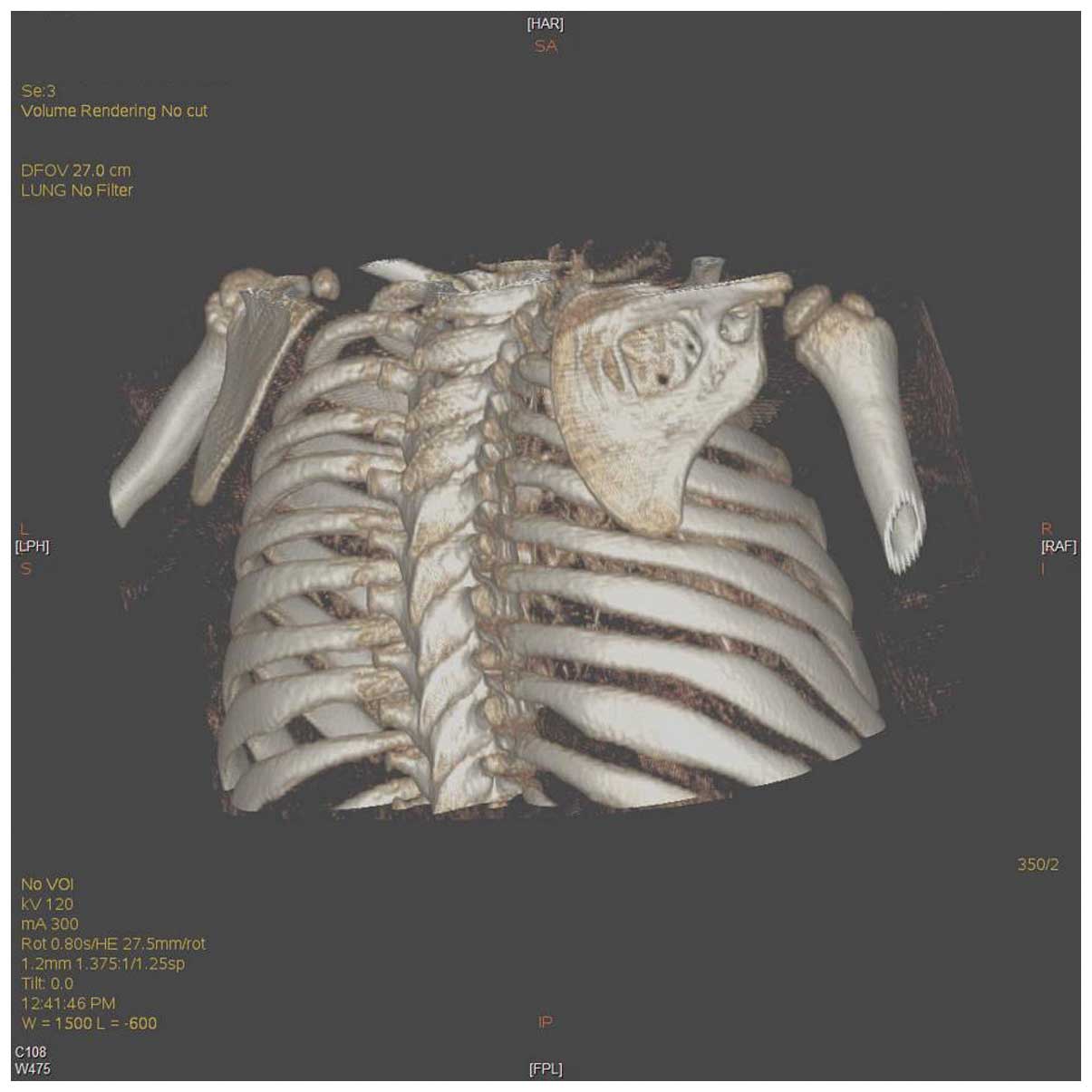
right scapula (Figs. 2–3). In addition, CT imaging revealed a soft
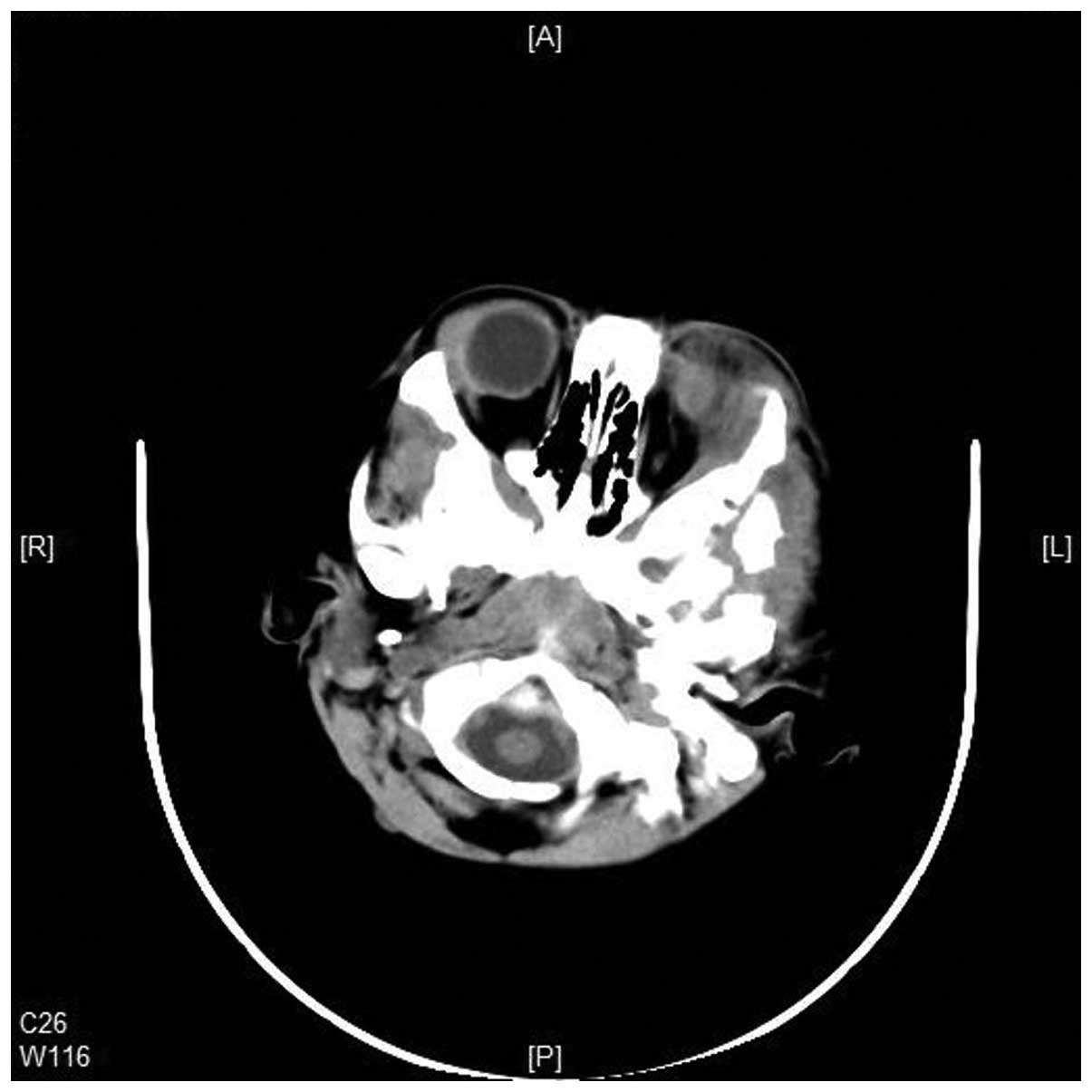
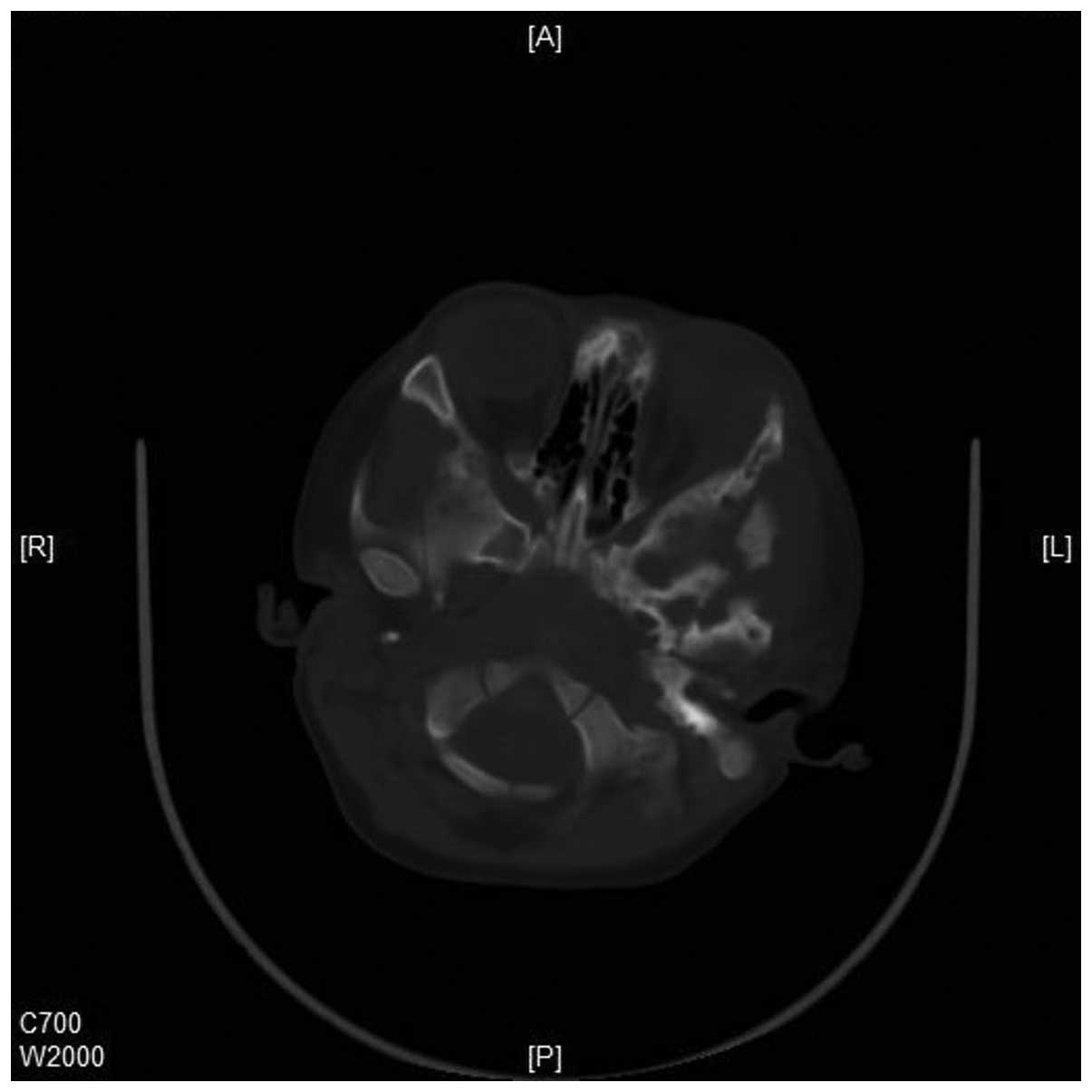
tissue mass in the left posterior orbit (Figs. 4 and 5). Increased interstitial markings, with
reticular patterns in the lungs (Fig.
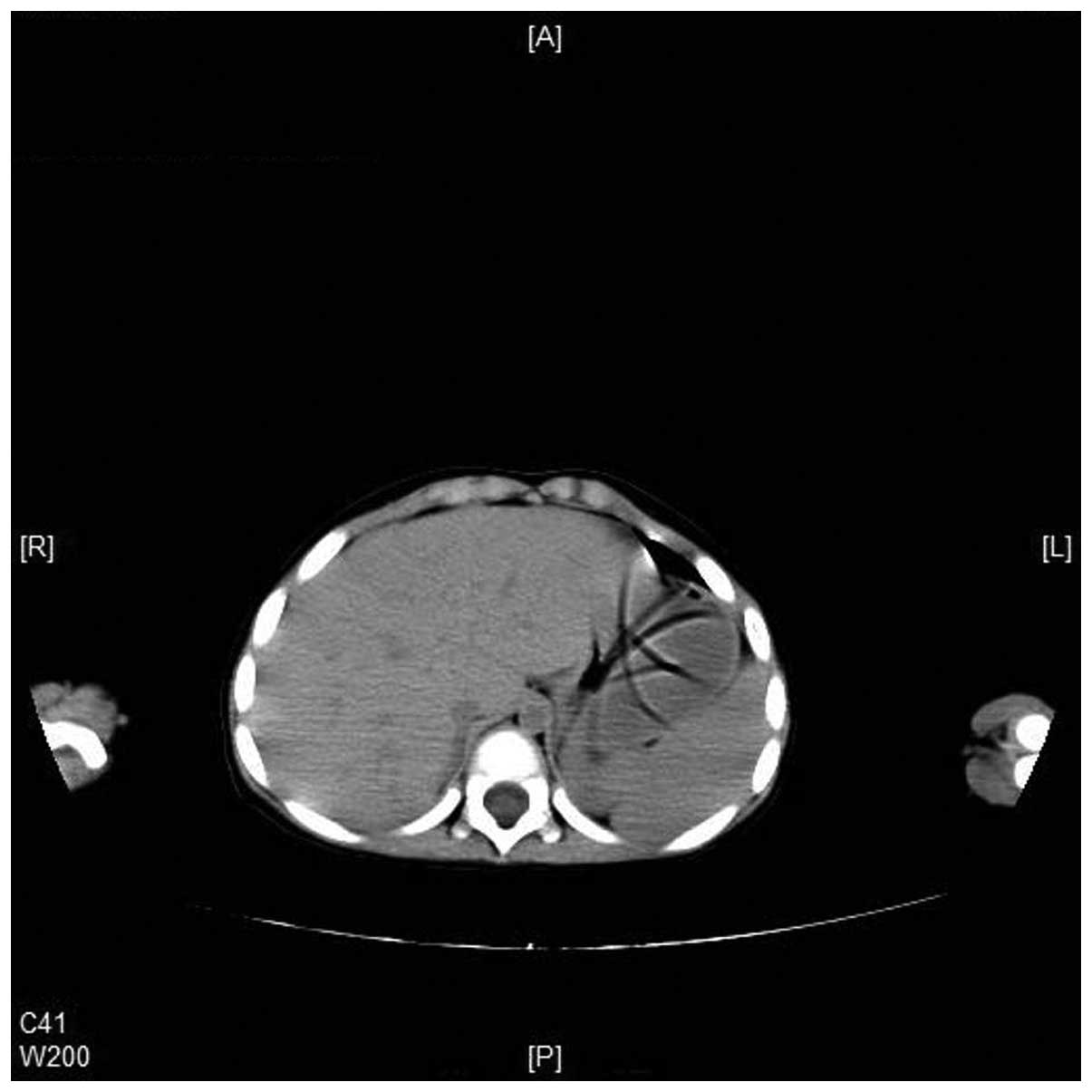
6), and enlargement of spleen (Fig.
7) were also observed in the CT scans. A complete blood count
from the patient revealed a hemoglobin level of 112 g/l, a white
blood cell count of 6.1×109/l and a platelet count of
456×109/l. Anaplastic lymphoma kinase, liver and kidney
function tests (alanine aminotransferase, 14 U/l; aspartate
aminotransferase, 22 U/l; creatinine 1.0 mg/dl) were normal. A skin
biopsy was performed and the results confirmed the diagnosis of
LCH. Immunohistochemical staining produced the following results:
CD1a, +++; fascin, +++; S-100, +++; Kp-1, sparsely +;
phosphoglucomutase-1, -; Ki-67, >20%+.
The patient subsequently received a chemotherapy
regimen consisting of vindesine, etoposide and prednisone (VEP
regimen). Following one cycle of chemotherapy, the patient's
temperature returned to normal, with rapid improvement observed
with regard to the dermatitis and otorrhea. In addition, the mass
on the scalp decreased in size. The VEP regimen was then alternated
with a VCP regimen, consisting of vindesine, cyclophosphamide and
prednisone. After 12 cycles (one cycle every ∼4 weeks), the patient
was shifted to vinblastine therapy, which was applied every two
weeks for one year, due to the major residual tumor burden. With
completion of the therapy for ∼2.5 years, the patient continues to
undergo follow-up and no evidence of active disease has been
observed.
Discussion
LCH is a rare disorder associated with a wide
spectrum of presenting symptoms and variable clinical behavior and
prognosis. Previously, the disease was separated into three
classifications, including eosinophilic granuloma,
Hand-Schüller-Christian and Letterer-Siwe (9). In 1990, a stratification system was
adopted by the LCH Study Group that classified the disease into
single system LCH and multisystem LCH (MS-LCH) (10). The former can be further divided into
single site (unifocal bone, skin or lymph node) and multiple site
(multifocal bone or lymph nodes) forms. MS-LCH is defined as having
more than two organ systems involved at the time of diagnosis. In
addition, MS-LCH can be subdivided into low-risk and high-risk
forms. Low-risk patients have no involvement of high-risk organs,
including the liver, lungs, spleen and hematopoietic system
(11). However, high-risk patients
have one or more of these organs involved (12). In the case of the present study, the
patient presented with lungs and spleen involvement; thus, was
classified in the high-risk group.
The diagnosis of LCH is based on histopathological
examination of the biopsy specimens. Pathologically, LCH is
characterized by multinucleated Langerhans' cells, histiocytes and
eosinophils, although the hallmark cell is the Langerhans cell
histiocyte. The gold standard for the diagnosis of the disease
requires the presence of Birbeck granules on electron microscopy
examination (13); however, in
certain cases, immunohistochemistry can play a fundamental role in
establishing a diagnosis. For example, a diagnosis can be
established with the use of CD1a, S100 and/or CD45 immunostaining
on histopathological specimens (14). Previously, a highly specific and
sensitive monoclonal antibody against CD207 (langerin) has become
commercially available. This protein appears to be important for
the formation of the Birbeck-Broadbent granules (15–17). In
the present study, a final diagnosis of LCH was confirmed based on
the histopathological observations and immunohistochemical staining
of the lesion cells with CD1a and S-100.
The etiology of LCH, which is characterized by a
clonal proliferation of histiocyte-like cells, is controversial.
There remains uncertainty regarding whether LCH is a neoplastic or
inflammatory disorder. LCH has been described as a neoplastic
process due to the monoclonal proliferation of Langerhans cells
(18). However, there is also the
possibility that LCH is a reactive inflammatory disorder resulting
from a dysregulation of the immune system (18). Previously, a number of findings with
regard to the molecular aspects of the disease suggested the
possibility that LCH may be the result of an immune dysregulation
(10,19–21). For
example, Langerhans cell histiocytes are known to release a number
of inflammatory chemokines, including C-C chemokine receptor
(CCR)1, CCR2, CCR5, CCR6, C-X-C chemokine receptor (CXCR)1, CXCR4,
CCR7, chemokine (C-C motif) ligand (CCL)20/macrophage inflammatory
protein (MIP)-3α, CCL2/ monocyte chemoattractant protein (MCP)-1,
CCL3/MIP-1α, CCL4/MCP-4α, CCL5/RANTES, chemokine (C-X-C motif)
ligand (CXCL8/interleukin (IL)-8 and CXCL10/IL-10. These chemokines
are important for the recruitment of circulating immature dendritic
cells, as well other immune cell types, such as T lymphocytes,
macrophages and eosinophils. Subsequently, the recruited cells
produce a number of additional cytokines, including IL-1, IL-3,
IL-4, IL-5, IL-8, IL-10, granulocyte-macrophage colony-stimulating
factor, tumor necrosis factor-α, transforming growth factor-β and
leukemia inhibitory factor. This particular immunological setting,
known as a ʻcytokine stormʼ, in which autocrine and paracrine
stimulation is established among the cells, can explain a number of
clinical symptoms, including fever and failure to thrive (22). By contrast, numerous genetic
abnormalities, including the loss of heterozygosity, damage to the
chromosomes and damage to genes (such as BRAF mutation), have been
detected in LCH, which supports the possible clonal-expansive
nature of the disease (22). These
two pathways are currently under investigation with the aim to
identify possible targets for the molecular therapy of LCH
(10,14,15,19–21,23–27). One
of the most exciting findings that may have therapeutical potential
is the presence of the BRAF V600E mutation in LCH. Previous studies
have demonstrated that BRAF V600E mutations are present in 50–60%
of patients with LCH (27,28). The LCH patients with a BRAF V600E
mutation respond to BRAF V600E inhibition. Haroche et al
reported a treatment protocol using vemurafenib (a BRAF inhibitor)
in three patients with multisystemic and refractory Erdheim-Chester
disease who carried the BRAF V600E mutation; two patients also had
skin or lymph node LCH involvement. For all the patients,
vemurafenib treatment resulted in a substantial and rapid clinical
and biological improvement (29).
However, more clinical trials are required to optimize the
risk/benefit ratio of BRAF V600E inhibition in children.
Currently, the treatment for LCH with multisystem
involvement is controversial. For patients with a multisystem
disease, systemic multiagent chemotherapy is recommended. The most
common chemotherapeutic agents are vinblastine, prednisone,
etoposide and methotrexate, applied in various combinations
(30). To date, a vinblastine- or
etoposide-based regimen is most common for the treatment of LCH
(31). However, severe and
refractory LCH patients, particularly those with a life-threatening
disease, may benefit from other therapies, including monoclonal
antibodies that target CD1a, CD207 or CD52, specific cytokine
inhibitors, 2-chlorodeoxyadenosine, BRAF V600E inhibition and bone
marrow transplantation (28–32). In the present study, the patient was
found to have a multivisceral form of the disease; thus, the VEP
and VCP regimens were selected and applied for a year, after which
the patient received vincristine therapy every two weeks for a
year.
LCH is a rare proliferative disease of
histiocyte-like cells that most commonly affects individuals in
childhood. Diagnosis of LCH should be based on the synthetical
analysis of clinical presentations, in addition to features of
imaging and histopathology. Although certain cases regress
spontaneously, other cases require systemic chemotherapy. The
present study reported a rare infant case of LCH presenting with
multisystem involvement, including that of the bone, skin, orbit,
spleen and lungs. A vindesine- and prednisone-based regimen was
selected for treatment and the patient was shown to progress well.
However, the patient continues to undergo close follow-up to assess
for signs of recurrence.
References
|
1
|
Satter EK and High WA: Langerhans cell
histiocytosis: A review of the current recommendations of the
Histiocyte Society. Pediatr Dermatol. 25:291–295. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Favara BE, McCarthy RC and Mierau GW:
Histiocytosis X. Hum Pathol. 14:663–676. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lieberman PH, Jones CR, Steinman RM, et
al: Langerhans cell (eosinophilic) granulomatosis. A
clinicopathologic study encompassing 50 years. Am J Surg Pathol.
20:519–552. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Egeler RM and D'Angio GJ: Langerhans cell
histiocytosis. J Pediatr. 127:1–11. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schulze J, Kitz R, Gruttner HP, et al:
Severe isolated pulmonary Langerhans cell histiocytosis in a
6-year-old girl. Eur J Pediatr. 163:320–322. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lajolo C, Campisi G, Deli G, et al:
Langerhans's cell histiocytosis in old subjects: Two rare case
reports and review of the literature. Gerodontology.
29:e1207–e1214. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Minkov M, Prosch H, Steiner M, et al:
Langerhans cell histiocytosis in neonates. Pediatr Blood Cancer.
45:802–807. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
McClain KL: Langerhans cell histiocytosis:
what is the orphan telling us? Hematology (Am Soc Hematol Educ
Program). 84–87. 2004.
|
|
9
|
Isaacs H Jr: Fetal and neonatal
histiocytoses. Pediatr Blood Cancer. 47:123–129. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Abla O, Egeler RM and Weitzman S:
Langerhans cell histiocytosis: Current concepts and treatments.
Cancer Treat Rev. 36:354–359. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Allen CE and McClain KL: Langerhans cell
histiocytosis: a review of past, current and future therapies.
Drugs Today (Barc). 43:627–643. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lau EG, Stepenaskie S, Moran R, et al:
ʻBlueberry muffinʼ rash and large right thigh mass: a unique
presentation of Langerhans cell histiocytosis. Dermatol Online J.
19:185682013.PubMed/NCBI
|
|
13
|
Herwig MC, Wojno T, Zhang Q and
Grossniklaus HE: Langerhans cell histiocytosis of the orbit: Five
clinicopathologic cases and review of the literature. Surv
Opthalmol. 58:330–340. 2013. View Article : Google Scholar
|
|
14
|
Kasper EM, Aguirre-Padilla DH, Alter RY,
et al: Histiocytosis X: Characteristics, behavior and treatments as
illustrated in a case series. Surg Neurol Int. 2:572011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kairouz S, Hashash J, Kabbara W, et al:
Dendritic cell neoplasms: an overview. Am J Hematol. 82:924–928.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Takahashi K, Harada M, Kimoto M and Kondo
F: Diagnostic confirmation of Langerhans cell histiocytosis of the
jaws with CD1a immunostaining: a case report. J Oral Maxillofac
Surg. 61:118–122. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Valladeau J, Dezutter-Dambuyant C and
Saeland S: Langerin/CD207 sheds light on formation of birbeck
granules and their possible function in Langerhans cells. Immunol
Res. 28:93–107. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rizzo FM, Cives M, Simone V, et al: New
insights into the molecular pathogenesis of langerhans cell
histiocytosis. Oncologist. 19:151–163. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Egeler RM, van Halteren AG, Hogendoorn PC,
et al: Langerhans cell histiocytosis: fascinating dynamics of the
dendritic cell-macrophage lineage. Immunol Rev. 234:213–232. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Allen CE, Li L, Peters TL, et al:
Cell-specific gene expression in Langerhans cell histiocytosis
lesions reveals a distinct profile compared with epidermal
Langerhans cells. J Immunol. 184:4557–4567. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Annels NE, Da Costa CE, Prins F, et al:
Aberrant chemokine receptor expression and chemokine production by
Langerhans cells underlies the pathogenesis of Langerhans cell
histiocytosis. J Exp Med. 197:1385–1390. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hicks J and Flaitz CM: Langerhans cell
histiocytosis: current insights in a molecular age with emphasis on
clinical oral and maxillofacial pathology practice. Oral Surg Oral
Med Oral Pathol Oral Radiol Endod. 100:(2 Suppl). 42–66. 2005.
View Article : Google Scholar
|
|
23
|
Bechan GI, Egeler RM and Arceci RJ:
Biology of Langerhans cells and Langerhans cell histiocytosis. Int
Rev Cytol. 254:1–43. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
De Filippi P, Badulli C, Cuccia M, et al:
Specific polymorphisms of cytokine genes are associated with
different risks to develop single-system or multi-system childhood
Langerhans cell histiocytosis. Br J Haematol. 132:784–787. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Egeler RM, Annels NE and Hogendoorn PC:
Langerhans cell histiocytosis: a pathologic combination of
oncogenesis and immune dysregulation. Pediatr Blood Cancer.
42:401–403. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chikwava KR, Hunt JL, Mantha GS, et al:
Analysis of loss of heterozygosity in single-system and multisystem
Langerhans' cell histiocytosis. Pediatr Dev Pathol. 10:18–24. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Badalian-Very G, Vergilio JA, Degar BA, et
al: Recurrent BRAF mutations in Langerhans cell histiocytosis.
Blood. 116:1919–1923. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sahm F, Capper D, Preusser M, et al:
BRAFV600E mutant protein is expressed in cells of variable
maturation in Langerhans cell histiocytosis. Blood. 120:e28–e34.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Haroche J, Cohen-Aubart F, Emile JF, et
al: Dramatic efficacy of vemurafenib in both multisystemic and
refractory Erdheim-Chester disease and Langerhans cell
histiocytosis harboring the BRAF V600E mutation. Blood.
121:1495–1500. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Minkov M: Multisystem Langerhans cell
histiocytosis in children: current treatment and future directions.
Paediatr Drugs. 13:75–86. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Behrman RE, Kliegman RM and Jenson HB:
Class 1 histiocytosesNelson Textbook of Pediatrics. 16th. W.B.
Saunders; Philadelphia, PA: pp. 1570–1572. 2000
|
|
32
|
Jordan MB, McClain KL, Yan X, Hicks J and
Jaffe R: Anti-CD52 antibody, alemtuzumab, binds to Langerhans cells
in Langerhans cell histiocytosis. Pediatr Blood Cancer. 44:251–254.
2005. View Article : Google Scholar : PubMed/NCBI
|