Introduction
Insulin resistance is the primary mechanism
underlying hyperglycemia in critically ill patients (1–4). Insulin
resistance and the resulting hyperglycemia seriously influence the
prognosis of critical patients, aggravating the disease and
increasing the risk of complications and mortality (5–8).
Propofol (2,6-diisopropylphenol) is the most common intravenous
anesthetic agent used to narcotize or mitigate pain in critically
ill patients. Recently, Yasuda et al (9) investigated the effects of propofol on
insulin sensitivity in rats. The results revealed that anesthesia
with propofol induced systemic insulin resistance through
decreasing insulin-stimulated glucose uptake in the skeletal and
heart muscle, and attenuating the insulin-mediated suppression of
hepatic glucose output.
However, little information is available with regard
to the effect of propofol alone on the insulin signaling pathway
and insulin resistance, excluding the effects of other factors,
such as surgical stress and the presence of fat-soluble carriers.
Thus, whether propofol aggravates insulin resistance in critically
ill patients and whether the infusion of propofol is safe in
critical patients with insulin resistance remain unknown.
The liver is the major target organ of insulin, and
the most important organ involved in the regulation of glucolipid
metabolism (10). In the present
study, the effects of propofol on insulin resistance in primary
mouse hepatocytes were examined with the aim to investigate the
molecular mechanisms underlying the effect of propofol on insulin
resistance.
The research results may provide a scientific basis
for the targeted prevention and treatment of insulin resistance in
critically ill patients, and guide the selection of appropriate
anesthetic methods and drugs clinically.
Materials and methods
Reagents
Clinical propofol injections contain numerous
auxiliary materials, including fats, soybean oils, purified
lecithin, glycerin and oleic acid, and high fat can induce insulin
resistance (11–13). In order to avoid the interference of
auxiliary materials in the propofol injection, propofol with a high
purity (97%; Sigma-Aldrich, St. Louis, MO, USA) was selected. Due
to the hydrophobicity of propofol, 0.1% (final concentration)
dimethyl sulfoxide (DMSO) was used as a solvent. Lithium chloride
(LiCl) was used in the experiment to determine whether GSK-3β is a
target of propofol. LiCl, tumor necrosis factor (TNF)-α and DMSO
were purchased from Sigma-Aldrich. TNF-α was used to induce insulin
resistance in primary mouse hepatocytes (14–16). The
culture reagents were purchased from Invitrogen Life Technologies
(Carlsbad, CA, USA), and the reagents used for SDS-PAGE were
obtained from Bio-Rad Laboratories, Inc. (Hercules, CA, USA).
Antibodies against Akt, phosphorylated (p) Akt (Ser473), glycogen
synthase kinase-3β (GSK-3β) and p-GSK-3β (Ser9) were purchased from
Cell Signaling Technology, Inc. (Danvers, MA, USA).
Animals
A total of 10 male C57BL/6J mice (age, 8 weeks;
weight, 22–32 g) were provided by Peking University Health Science
Center (Beijing, China). A single mouse provided between
5×107 and 5×108 primary hepatocytes. Animal
procedures were performed in accordance with the National
Institutes of Health Animal Care and Use Guidelines (17), and animal experimental protocols were
approved by the Animal Ethics Committee of Zhujiang Hospital
(Guangzhou, China).
Isolation of mouse primary
hepatocytes
Primary hepatocytes were isolated using a two-step
collagenase perfusion method [0.2 mg/ml type IV collagenase
(Sigma-Aldrich) in Hank's balanced salt solution], as described
previously (18,19). The hepatocytes were collected by
centrifugation at 120 × g for 8 min. Immediately following
harvesting, the cells were suspended in prewarmed William's E
medium (Sigma-Aldrich) that was supplemented with 10% fetal bovine
serum, 20 ng/ml dexamethasone (Sigma-Aldrich), insulin (5 mg/l),
transferrin (5 mg/l), sodium selenate (5 µg/l; Sigma-Aldrich) and
10 µg/ml gentamicin (Invitrogen Life Technologies). The hepatocytes
were plated in collagen-coated 25-cm2 flasks at a
density of 1×106 cells/flask and would be used as a
control in the following experiment.
Western blot analysis
Cell lysates (15–30 µg protein) were separated by
10% SDS-PAGE and transferred to polyvinyldifluoride membranes
(Millipore Corporation, Billerica, MA, USA), after which the
membranes were blocked with 5% nonfat dry milk. Subsequently, the
membranes were probed with antibodies against Akt, p-Akt, GSK and
p-GSK at 4°C overnight. The blots were also probed with a β-actin
antibody to ensure that approximately equal amounts of protein were
loaded. Next, the blots were incubated with a horseradish
peroxidase-conjugated anti-IgG secondary antibody, which was
followed by detection with enhanced chemiluminescence (Millipore
Corporation).
Analysis of the glycogen content
Glycogen levels were measured in the cells for 3 h
in the presence of 10 nmol/l insulin (USBio, Salem, MA, USA) using
a glycogen assay kit (Biovision, Inc., Milpitas, CA, USA).
Cell viability assay
A
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
tetrazole (MTT) reduction assay was used to asses cell viability.
Mouse primary hepatocytes were plated in 24-well plates
(3×104 cells per well). After incubation for 24 h, the
cells were treated with increasing concentrations of propofol for
24 h. MTT (0.5 mg/ml; Sigma-Aldrich) was then added to each well
(200 µl/well). After additional incubation for 4 h, MTT solution
was discarded and 200 µl DMSO (Amresco LLC, Solon, OH, USA) was
added and the plates were shaken gently. The absorbance was
measured using an ELISA reader at a wavelength of 490 nm.
Statistical analysis
Data represent the mean of duplicate samples from
three separately performed experiments, and the results are
expressed as the mean ± standard deviation. Statistical analysis
was performed with SPSS statistical software (version 19.0; IBM
SPSS, Armonk, NY, USA). Differences between two groups were
analyzed for statistical significance using the Student's
t-test, while one-way analysis of variance, followed by
Tukey's test, were used to compare the differences among >2
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
Effects of propofol on the cell
viability of mouse primary hepatocytes
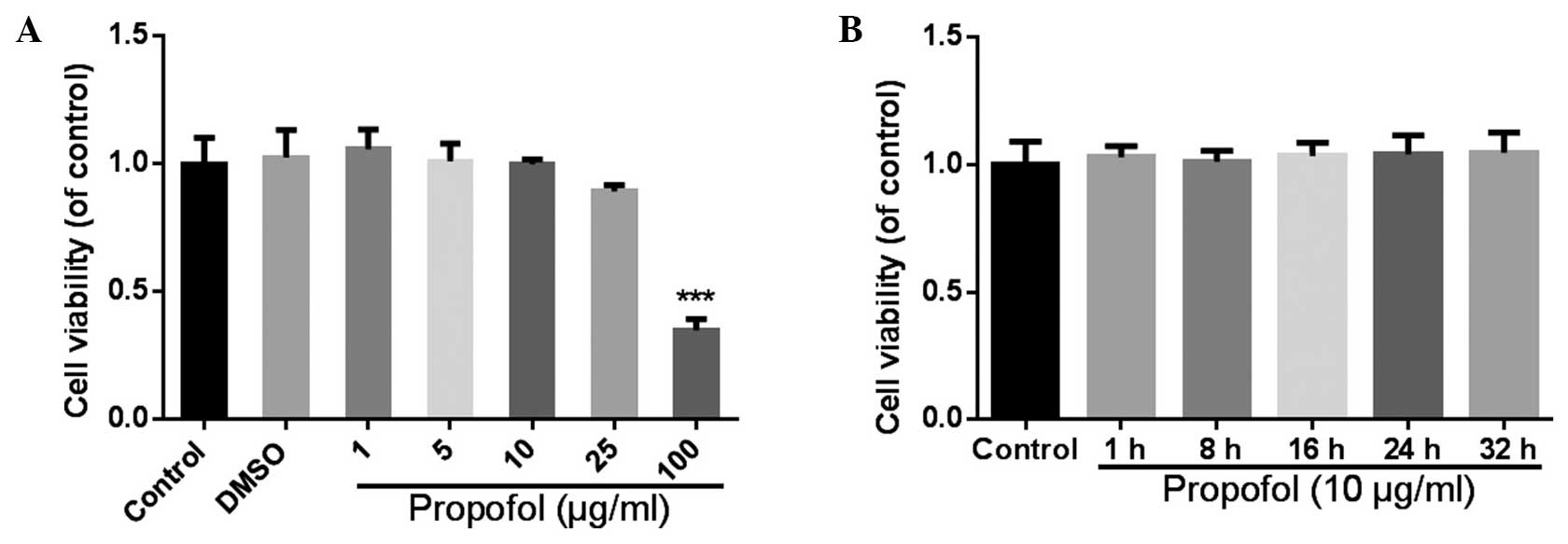
Cell viability of the primary mouse hepatocytes was
detected with a MTT assay, subsequent to the hepatocytes being
treated with different concentrations of propofol for 24 h.
Propofol at concentrations between 1 and 25 µg/ml was shown to have
no significant effect on the cell viability (Fig. 1A); however, when the final
concentration of propofol reached 100 µg/ml, the cell viability
decreased to 35±5% of the control (P<0.001). Thus, a 10-µg/ml
concentration of propofol was selected to use in the subsequent
experiments, since minimal effects were observed on the cell
viability compared with higher doses, and use of this concentration
had been previously reported (20,21). In
the following experiment, the effect of propofol at different
culture periods (1–32 h) was analyzed. The results revealed no
statistically significant differences in the viability of the
hepatocytes following treatment with 10 µg/ml propofol between 1
and 32 h (P>0.05; Fig. 1B). From
these results, the dose of propofol selected was demonstrated to
not affect the viability of the hepatocyptes during the culture
period of the subsequent assays.
Propofol induces insulin resistance in
mouse primary hepatocytes
Primary mouse hepatocytes were treated with 10 µg/ml
propofol for 24 h, after which the cells were assayed with western
blot analyses to detect the protein expression levels of components
of the phosphoinositide 3-kinase (PI3K)/Akt/GSK-3β signaling
pathway. In addition, a glycogen assay kit was used to detect the
level of glycogen synthesis. Following treatment with propofol for
24 h, the phosphorylation levels of Akt (Ser473) and GSK-3β (Ser9)
were found to decrease (Fig. 2A–C),
and the rate of glycogen synthesis had reduced (Fig. 2D). Hepatocytes were simultaneously
treated with propofol (final concentration, 10 µg/ml) and LiCl
(final concentration, 20 µmol/l) for 24 h. No statistically
significant difference was observed in the level of glycogen
synthesis or the ratio of pGSK-3β (Ser9)/GSK-3β between the
propofol + LiCl and DMSO groups (P>0.05). However, a
statistically significant difference was observed in the ratio of
pAkt (Ser473)/Akt between the propofol + LiCl and DMSO groups
(P<0.05), and a statistically significant difference was
observed in the ratio of pAkt (Ser473)/Akt between the propofol +
LiCl and propofol groups (P<0.05). Furthermore, a statistically
significant difference was observed between pAkt (Ser473) and Akt
expression levels, as well as between propofol alone and propofol +
LiCl (P<0.05).
 | Figure 2.(A) Western blot analysis showing the
protein expression levels of pAkt (Ser473), Akt, pGSK-3β (Ser9) and
GSK-3β, where β-actin was used as an internal control. (B) Propofol
inhibits pAkt (Ser473)/Akt expression in mouse primary hepatocytes.
**P<0.01, propofol vs. DMSO group; *P<0.05, propofol + LiCl
vs. DMSO group. (C) Propofol inhibits pGSK-3β (Ser9)/GSK-3β
expression in mouse primary hepatocytes. **P<0.01, propofol vs.
DMSO group. (D) Glycogen levels were measured in mouse primary
hepatocytes using a glycogen assay kit. Propofol inhibits glycogen
synthesis in mouse primary hepatocytes. *P<0.05, propofol vs.
DMSO group. LiCl, lithium chloride; DMSO, dimethylsulfoxide; GSK,
glycogen synthase kinase; n.s., not significant. |
TNF-α inhibits the PI3K/Akt/GSK-3β
signaling pathway and glycogen synthesis in mouse primary
hepatocytes
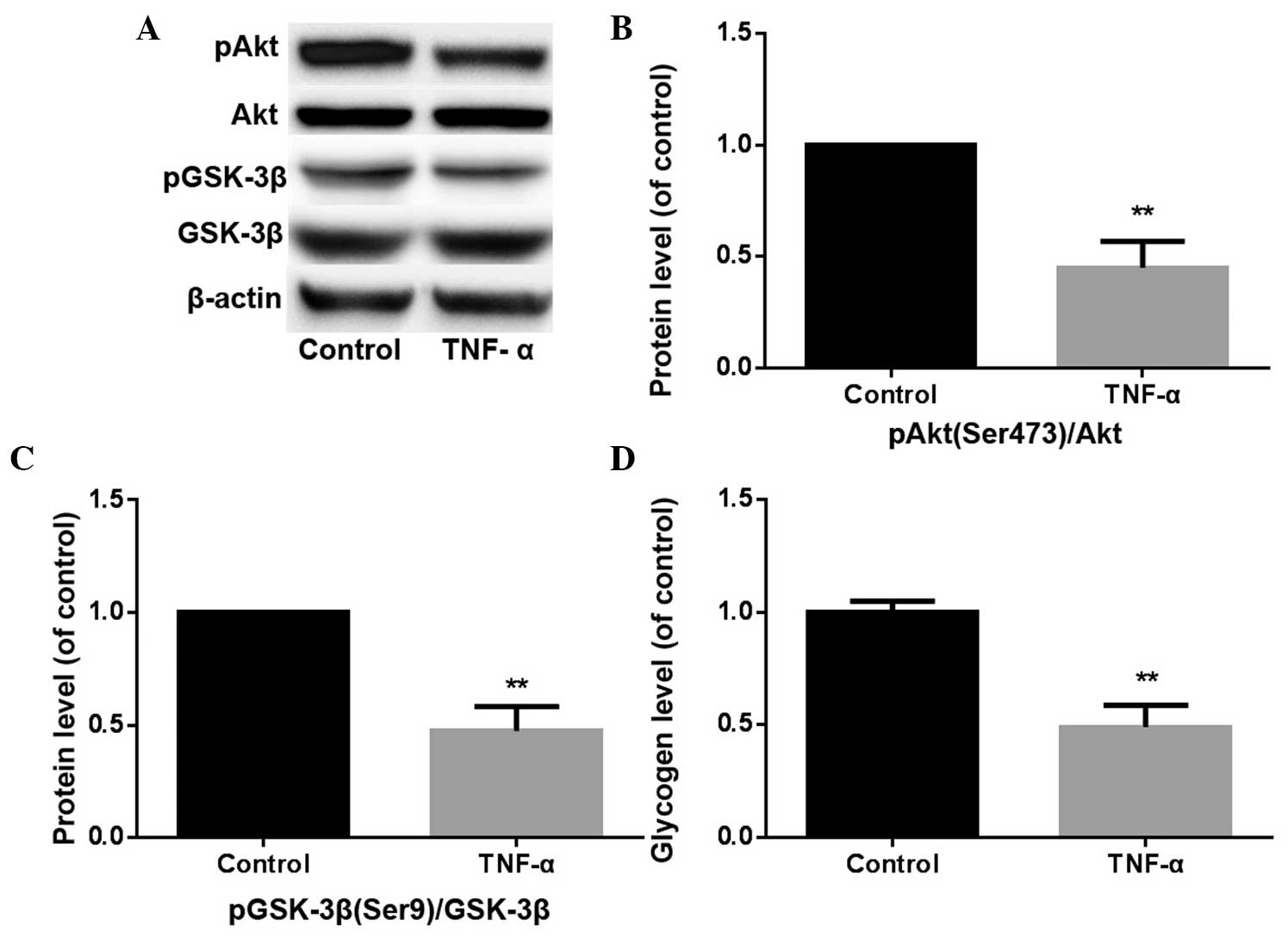
Following treatment with TNF-α for 24 h, the
expression levels of pAkt (Ser473)/Akt decreased to 45±12% of that
in the control group (P<0.01), while the pGSK-3β (Ser9)/GSK-3β
expression levels decreased to 47±11% of that in the control group
(P<0.01). In addition, the level of glycogen synthesis declined
to 49±10% of that in the control group (P<0.01; Fig. 3). Therefore, TNF-α was shown to mimic
the effects of propofol.
Pretreatment with propofol alleviates
the inhibition of TNF-α on the PI3K/Akt/GSK-3β signaling pathway
and glycogen synthesis in mouse primary hepatocytes
Hepatocytes were treated with 10 µg/ml propofol for
6 h, and subsequently treated with 10 ng/ml TNF-α for 24 h. The
results revealed that the expression levels of pAkt (Ser473)/Akt
and pGSK-3β (Ser9)/GSK-3β, and the glycogen content in the propofol
pretreatment group, were higher compared with those in the DMSO +
TNF-α group (P<0.001, P<0.01 and P<0.05, respectively;
Fig. 4).
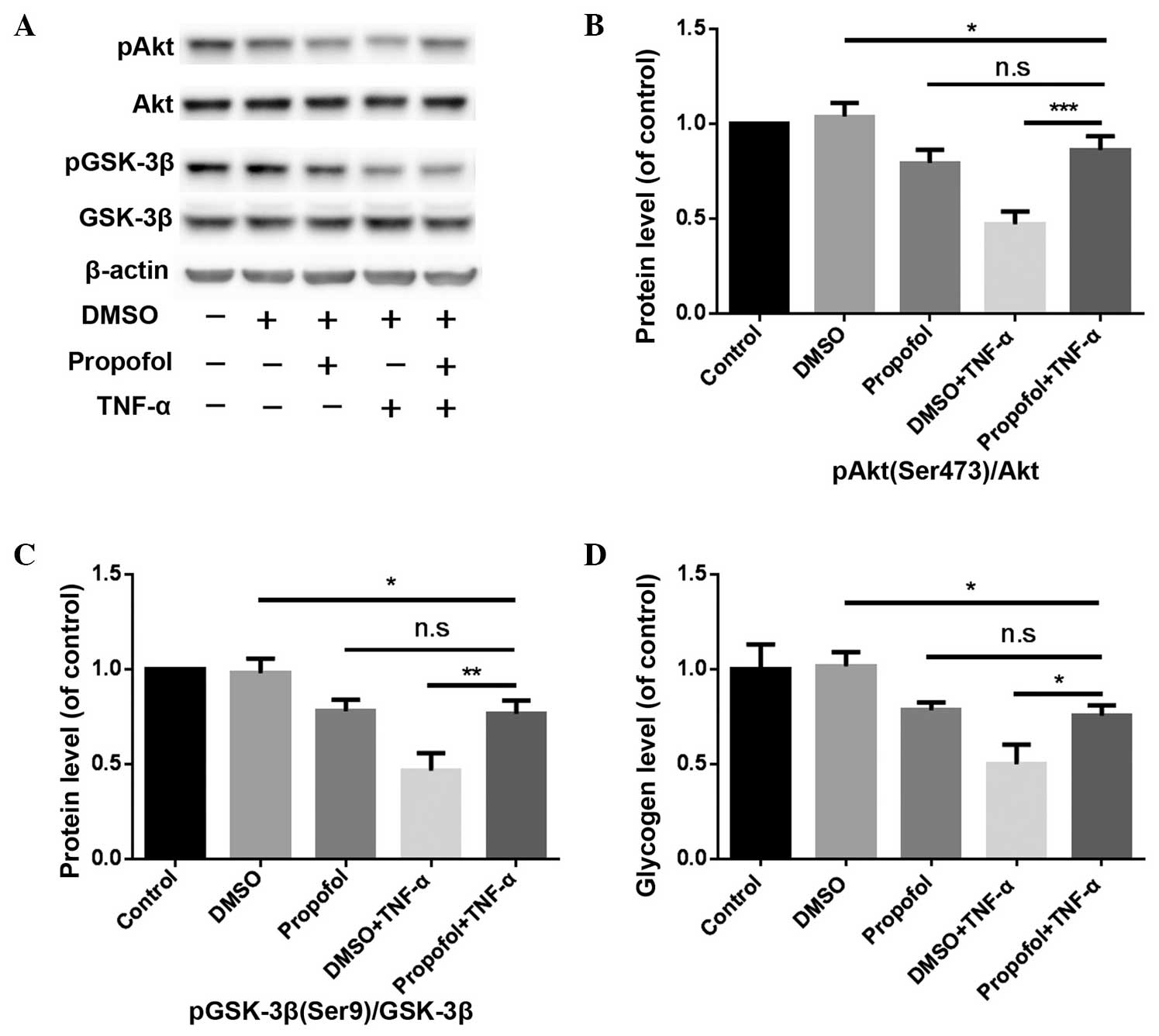 | Figure 4.Pretreatment with propofol alleviates
the inhibition of TNF-α on the PI3K/Akt/GSK-3β signaling pathway
and glycogen synthesis in mouse primary hepatocytes. (A) Western
blot analysis showing the protein expression levels of pAkt
(Ser473), Akt, pGSK-3β (Ser9) and GSK-3β, where β-actin was used as
an internal control. (B) Effect of the different treatments on pAkt
(Ser473)/Akt protein expression levels. ***P<0.001, propofol +
TNF-α vs. DMSO + TNF-α groups. (C) Effect of the different
treatments on pGSK-3β (Ser9)/GSK-3β protein expression levels.
**P<0.01, propofol + TNF-α group vs. DMSO + TNF-α group. (D)
Glycogen levels were measured in mouse primary hepatocytes using a
glycogen assay kit. *P<0.05, propofol + TNF-α group vs. DMSO +
TNF-α group. TNF, tumor necrosis factor; PI3K, phosphoinositide
3-kinase; GSK, glycogen synthase kinase; DMSO, dimethylsulfoxide;
n.s., not significant. |
Discussion
Insulin resistance is a physiological state that is
characterized by the failure to suppress glycogenolysis or hepatic
glucose production (22). The
blockage or weakening of insulin signal transduction, which can be
induced by multiple factors, is the main pathogenetic mechanism
underlying insulin resistance (23).
The PI3K-Akt signaling pathway is a classic insulin signal
transduction pathway (24), and all
the factors that directly or indirectly influence this pathway are
able to induce insulin resistance. In the present study, propofol
was demonstrated to significantly reduce the phosphorylation levels
of Akt (Ser473) and GSK-3β (Ser9), which subsequently blocked the
PI3K/Akt/GSK-3β signaling pathway and inhibited glycogen synthesis
in primary mouse hepatocytes. These results indicated that propofol
induced insulin resistance in primary mouse hepatocytes.
Lithium selectively inhibits GSK-3β activity
(25,26). As shown in Fig. 2, there were no statistically
significant differences in the glycogen synthesis level (P>0.05)
or the GSK-3β (Ser9) phosphorylation level (P>0.05) when
comparing the group treated with propofol and lithium and the
control group. However, the phosphorylation level of Akt (Ser473)
was significantly different (P<0.05) between the two groups.
These results indicate that LiCl counteracts the inhibitory effect
of propofol on glycogen synthesis in primary mouse hepatocytes. In
addition, the results demonstrate that the inhibition of GSK-3β
(Ser9) phosphorylation is a critical step in the inhibitory effect
of propofol on glycogen synthesis in primary mouse hepatocytes.
However, LiCl was unable to completely eliminate the inhibitory
effect of propofol on the PI3K/Akt/GSK-3β signaling pathway,
indicating that the target of propofol for the induction of insulin
resistance in primary mouse hepatocytes was upstream of GSK-3β.
TNF-α, which is mainly secreted by monocytes or
macrophages, is an important proinflammatory cytokine, and also a
key component in obesity and insulin resistance (27,28). In
the present study, TNF-α was shown to inhibit the PI3K/Akt/GSK-3β
signaling pathway and glycogen synthesis in primary mouse
hepatocytes. Furthermore, TNF-α induced insulin resistance in the
primary mouse hepatocytes. By this means, the cell model of insulin
resistance was successfully constructed.
As shown in Fig. 4,
the phosphorylation levels of Akt (Ser473) and GSK-3β (Ser9), as
well as the total level of glycogen synthesized, in the group
treated with propofol and TNF-α were higher compared with the
control group treated with TNF-α alone. These observations
indicated that pretreatment with propofol alleviated the inhibitory
effects of TNF-α on the PI3K/Akt/GSK-3β signaling pathway and
glycogen synthesis in primary mouse hepatocytes. Furthermore, the
present results indicated that propofol exerted a protective effect
on the insulin resistance of primary mouse hepatocytes induced by
TNF-α. Propofol administration alone was shown to inhibit the
PI3K/Akt/GSK-3β signaling pathway in primary mouse hepatocytes.
Notably, pretreatment with propofol was also shown to alleviate the
inhibition of TNF-α on the PI3K/Akt/GSK-3β signaling pathway in
primary mouse hepatocytes. These two seemingly contradictory
results indicate that the protective effect of propofol on
TNF-α-induced insulin resistance in primary mouse hepatocytes is
not achieved through a direct effect on the PI3K-Akt signaling
pathway.
Nuclear factor-κB (NF-κB) is widely distributed in
tissue cells (29). The
transcription factor plays an important role in cell signal
transduction and gene expression regulation, and is also the
critical nuclear factor involved in the initiation and regulation
of inflammation. In recent years, the inflammatory response
mediated by NF-κB is one focus of research into the mechanisms
underlying insulin resistance (30,31).
Following NF-κB activation, the transcription of inflammatory
factors, such as TNF-α, interleukin (IL)-1β and IL-6, is initiated
and regulated. These transcription factors serve as new activators
of NF-κB, and subsequently, a positive feedback loop of low
inflammation signaling is formed. As a result, insulin resistance
is generated or aggravated (29,32,33).
Previous studies have demonstrated that propofol inhibits NF-κB
activity in various tissues or cells (34–38).
Therefore, it was hypothesized that the protective effect of
propofol against TNF-α-induced insulin resistance in primary mouse
hepatocytes may be associated with the inhibitory effect of
propofol on the NF-κB signaling pathway.
In conclusion, propofol was demonstrated to induce
insulin resistance in primary mouse hepatocytes, while pretreatment
with propofol was shown to alleviate insulin resistance in primary
mouse hepatocytes induced by TNF-α. These results indicate that
propofol may alleviate insulin resistance in critically ill
patients; thus, the infusion of propofol in critically ill patients
may be clinically feasible.
Acknowledgements
This study was supported by grants from Shenzhen
Science and Technology Innovation Committee Project (no.
JCYJ20140403093211510) and Shenzhen Health and Family Planning
Commission Project (no. 201401074).
References
|
1
|
Fahy BG, Sheehy AM and Coursin DB: Glucose
control in the intensive care unit. Crit Care Med. 37:1769–1776.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gauglitz GG, Herndon DN and Jeschke MG:
Insulin resistance postburn: Underlying mechanisms and current
therapeutic strategies. J Burn Care Res. 29:683–694. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lipshutz AK and Gropper MA: Perioperative
glycemic control: An evidence-based review. Anesthesiology.
110:408–421. 2009.PubMed/NCBI
|
|
4
|
Coursin DB, Connery LE and Ketzler JT:
Perioperative diabetic and hyperglycemic management issues. Crit
Care Med. 32:(Suppl). S116–S125. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
van den Berghe G, Wouters P, Weekers F, et
al: Intensive insulin therapy in critically ill patients. N Engl J
Med. 345:1359–1367. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Krinsley JS: Association between
hyperglycemia and increased hospital mortality in a heterogeneous
population of critically ill patients. Mayo Clin Proc.
78:1471–1478. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Laird AM, Miller PR, Kilgo PD, Meredith JW
and Chang MC: Relationship of early hyperglycemia to mortality in
trauma patients. J Trauma. 56:1058–1062. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Deedwania P, Kosiborod M, Barrett E,
Ceriello A, Isley W, Mazzone T and Raskin P: American Heart
Association Diabetes Committee of the Council on Nutrition,
Physical Activity, and Metabolism: Hyperglycemia and acute coronary
syndrome: A scientific statement from the American Heart
Association Diabetes Committee of the Council on Nutrition,
Physical Activity, and Metabolism. Circulation. 117:1610–1619.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yasuda Y, Fukushima Y, Kaneki M and Martyn
JA: Anesthesia with propofol induces insulin resistance
systemically in skeletal and cardiac muscles and liver of rats.
Biochem Biophys Res Commun. 431:81–85. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Felig P and Wahren J: Influence of
endogenous insulin secretion on splanchnic glucose and amino acid
metabolism in man. J Clin Invest. 50:1702–1711. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pehmøller C, Brandt N, Birk JB, et al:
Exercise alleviates lipid-induced insulin resistance in human
skeletal muscle-signaling interaction at the level of TBC1 domain
family member 4. Diabetes. 61:2743–2752. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schenk S and Horowitz JF: Acute exercise
increases triglyceride synthesis in skeletal muscle and prevents
fatty acid-induced insulin resistance. J Clin Invest.
117:1690–1698. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Boden G, Lebed B, Schatz M, Homko C and
Lemieux S: Effects of acute changes of plasma free fatty acids on
intramyocellular fat content and insulin resistance in healthy
subjects. Diabetes. 50:1612–1617. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bastard JP, Maachi M, Lagathu C, Kim MJ,
Caron M, Vidal H, Capeau J and Feve B: Recent advances in the
relationship between obesity, inflammation and insulin resistance.
Eur Cytokine Netw. 17:4–12. 2006.PubMed/NCBI
|
|
15
|
Xu J, Kim HT, Ma Y, Zhao L, Zhai L,
Kokorina N, Wang P and Messina JL: Trauma and hemorrhage-induced
acute hepatic insulin resistance: Dominant role of tumor necrosis
factor-α. Endocrinology. 149:2369–2382. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Plomgaard P, Nielsen AR, Fischer CP,
Mortensen OH, Broholm C, Penkowa M, Krogh-Madsen R, Erikstrup C,
Lindegaard B, Petersen AM, et al: Associations between insulin
resistance and TNF-alpha in plasma, skeletal muscle and adipose
tissue in humans with and without type 2 diabetes. Diabetologia.
50:2562–2571. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shi XP, Zong A, Tao J and Wang L: Study of
instructive notions with respect to caring for laboratory animals.
Zhong Guo Yi Ke Da Xue Xue. 36:493. 2007.(In Chinese).
|
|
18
|
Seglen PO: Preparation of isolated rat
liver cells. Methods Cell Biol. 13:29–83. 1976.PubMed/NCBI
|
|
19
|
Casciano DA: Development and utilization
of primary hepatocyte culture systems to evaluate metabolism, DNA
binding, and DNA repair of xenobiotics. Drug Metab Rev. 32:1–13.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Smith C, McEwan AI, Jhaveri R, et al: The
interaction of fentanyl on the Cp50 of propofol for loss of
consciousness and skin incision. Anesthesiology. 81:820–828;
discussion 26A. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hsing CH, Chen YH, Chen CL, et al:
Anesthetic propofol causes glycogen synthase kinase-3β-regulated
lysosomal/mitochondrial apoptosis in macrophages. Anesthesiology.
116:868–881. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Muniyappa R, Lee S, Chen H and Quon MJ:
Current approaches for assessing insulin sensitivity and resistance
in vivo: Advantages, limitations, and appropriate usage. Am J
Physiol Endocrinol Metab. 294:E15–E26. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Morino S, Kondo T, Sasaki K, et al: Mild
electrical stimulation with heat shock ameliorates insulin
resistance via enhanced insulin signaling. PLoS One. 3:e40682008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu P, Cheng H, Roberts TM and Zhao JJ:
Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev
Drug Discov. 8:627–644. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hong M, Chen DC, Klein PS and Lee VM:
Lithium reduces tau phosphorylation by inhibition of glycogen
synthase kinase-3. J Biol Chem. 272:25326–25332. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ryves WJ and Harwood AJ: Lithium inhibits
glycogen synthase kinase-3 by competition for magnesium. Biochem
Biophys Res Commun. 280:720–725. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hotamisligil GS: Inflammatory pathways and
insulin action. Int J Obes Relat Metab Disord. 27:(Suppl 3).
S53–S55. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Stephens JM, Lee J and Pilch PF: Tumor
necrosis factor-alpha-induced insulin resistance in 3T3-L1
adipocytes is accompanied by a loss of insulin receptor substrate-1
and GLUT4 expression without a loss of insulin receptor-mediated
signal transduction. J Biol Chem. 272:971–976. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Baeuerle PA and Baltimore D: NF-kappa B:
Ten years after. Cell. 87:13–20. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li Y, Yan H, Zhang Z, Zhang G, Sun Y, Yu
P, Wang Y and Xu L: Andrographolide derivative AL-1 improves
insulin resistance through down-regulation of NF-κB signalling
pathway. Br J Pharmacol. 2015.(Epub ahead of print). View Article : Google Scholar
|
|
31
|
Zhou MS, Liu C, Tian R, Nishiyama A and
Raij L: Skeletal muscle insulin resistance in salt-sensitive
hypertension: Role of angiotensin II activation of NF-κB.
Cardiovasc Diabetol. 14:452015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tak PP and Firestein GS: NF-κB: A key role
in inflammatory diseases. J Clin Invest. 107:7–11. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gao Z, Hwang D, Bataille F, Lefevre M,
York D, Quon MJ and Ye J: Serine phosphorylation of insulin
receptor substrate 1 by inhibitor kappa B kinase complex. J Biol
Chem. 277:48115–48121. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Du QH, Xu YB, Zhang MY, Yun P and He CY:
Propofol induces apoptosis and increases gemcitabine sensitivity in
pancreatic cancer cells in vitro by inhibition of nuclear factor-κB
activity. World J Gastroenterol. 19:5485–5492. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ye HH, Wu KJ, Fei SJ, Zhang XW, Liu HX,
Zhang JL and Zhang YM: Propofol participates in gastric mucosal
protection through inhibiting the Toll-like receptor-4/nuclear
factor kappa-B signaling pathway. Clin Res Hepatol Gastroenterol.
37:e3–e15. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li Q, Zhang L, Han Y, Jiang Z and Wang Q:
Propofol reduces MMPs expression by inhibiting NF-κB activity in
human MDA-MB-231 cells. Biomed Pharmacother. 66:52–56. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hsing CH, Lin MC, Choi PC, et al:
Anesthetic propofol reduces endotoxic inflammation by inhibiting
reactive oxygen species-regulated Akt/IKKbeta/NF-κB signaling. PLoS
One. 6:e175982011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li H, Tan J, Zou Z, Huang CG and Shi XY:
Propofol post-conditioning protects against cardiomyocyte apoptosis
in hypoxia/reoxygenation injury by suppressing nuclear factor-kappa
B translocation via extracellular signal-regulated kinase
mitogen-activated protein kinase pathway. Eur J Anaesthesiol.
28:525–534. 2011. View Article : Google Scholar : PubMed/NCBI
|