Introduction
Doxorubicin (DOX) is one of the most widely used
anticancer drugs, due to its potent therapeutic effects on a
variety of cancer types, including leukemia, lymphomas and breast
cancer. However, the clinical use of DOX is limited by severe toxic
side-effects on the heart, potentially resulting in congestive
heart failure and dilated cardiomyopathy (1). Numerous studies have demonstrated that
reactive oxygen species (ROS) production has been implicated in the
cardiotoxicity of DOX, which ultimately results in endothelial
dysfunction (2,3) and cardiomyocyte apoptosis (4).
Transcription factors of the forkhead box, class O
(FoxO) family are crucial regulators of the cellular stress
response and promote the cellular antioxidant defense. FoxOs
stimulate the transcription of genes coding for antioxidant
proteins located in various subcellular compartments, such as in
mitochondria, including superoxide dismutase-2 and peroxiredoxins
3/5. In previous studies, resveratrol has been demonstrated to
protect PC12 cells against high glucose-induced neurotoxicity via
the PI3K/Akt/FoxO3a pathway. Various antioxidant pathways regulate
or protect the cellular response to oxidative stress. Several
antioxidants, including peroxiredoxins (Prxs), are components of a
ubiquitous thioredoxin-dependent antioxidant defense system, which
catalyzes ROS inactivation in mammalian cells (5–7).
Frequently, multiple mammalian Prxs (including Prx I to VI) coexist
in various intracellular locations in the same cell (8,9). These
act as scavengers of cellular H2O2 that is
released following stimulation with various growth factors during
apoptosis, oxidative stress or proliferation (8,9). In
particular, Prx III has been demonstrated to exhibit a protective
role in cisplatin- and gentamicin-induced apoptosis through a
mitochondria-dependent pathway (10). Previous studies reported that
overexpression of Prx III protects the mouse myocardium from
infarction (11), whereas depletion
of Prx III results in increased intracellular levels of
H2O2, sensitizing cells to apoptotic
signaling (12). The forkhead box
transcription factor FoxO3a is a key transcription factor for
resistance to oxidative stress. Chiribau et al (13) demonstrated that Prx III expression in
human cardiac fibroblasts was regulated by FoxO3a during oxidative
stress. The authors also identified specific DNA-binding elements
for FoxO3a in the Prx III promoter (13). The aim of the present study was to
examine whether oxidative stress is able to induce Prx III
expression in an injury model of DOX-induced H9c2 cells. In
addition, the study investigated whether Prx III expression is
regulated by FoxO3a in H9c2 cells.
Materials and methods
Materials
A methyl thiazolyl tetrazolium bromide (MTT) assay,
Hoechst 33258, DOX, H2O2 and
N-acetyl-L-cysteine (NAC) were purchased from Sigma-Aldrich (St.
Louis, MO, USA). All the medium components used in cell cultures
were purchased from Thermo Fisher Scientific (Waltham, MA, USA),
unless otherwise stated. H9c2 cardiac myocytes were obtained from
the Shanghai Cell Library of China (cells were originally from
ATCC, Manassas, VA, USA).
Cell culture
H9c2 cardiac myocytes were cultured in Dulbecco's
modified Eagle's medium (DMEM) supplemented with 10% fetal bovine
serum (FBS), 100 µg/ml streptomycin and 100 U/ml penicillin (all
from Gibco Life Technologies, Carlsbad, CA, USA) in a humidified 5%
CO2 atmosphere at 37°C. H9c2 cardiac myocytes were
passaged every 2 days, for 5–8 passages. Subsequently, the cells
were seeded at a density of 2×106 cells/dish in 100 mm
dishes with 10% calf serum, incubated for 24 h and then the medium
was changed to 0.5% FBS-supplemented DMEM for 24-h starvation.
MTT assay
The MTT assay is a standard method used to assess
cell viability. Prior to each experiment, H9c2 cardiac myocytes
(5,000 cells/well) were seeded in 96-well microtiter plates.
Following incubation with NAC for 60 min, the cells were treated
with 5 µM DOX and incubated for a further 24 h. Subsequently, 10 µl
MTT solution was added to each well, followed by incubation for 4 h
at 37°C. The absorbance was then measured at 470 nm and the values
were used to calculate the relative ratio of cell viability. Three
independent experiments were performed for each experimental
condition. The various experimental groups were as follows: Control
group, untreated H9c2 cells incubated in 0.5% FBS DMEM for 24 h;
DOX group, cells treated with 5 µM DOX for 24 h; DOX + NAC group,
cells treated with 1,000 µM NAC for 60 min prior to exposure to
DOX; and the H2O2 group, cells treated with
200 µM H2O2 for 12 h.
Assessment of cardiomyocyte cell
apoptosis
Apoptosis was analyzed by fluorescence microscopy
with the chromatin dye, Hoechst 33258. Following various
treatments, the cells were fixed in ice-cold 4% paraformaldehyde
dissolved in phosphate-buffered saline (PBS) at room temperature
for 20 min. Nonspecific binding was blocked using 5% normal goat
serum in 0.01 M PBS containing 0.3% Triton X-100. Next, the cells
were washed twice with PBS and incubated with 10 µg/ml Hoechst
33258 for 10 min at room temperature in the dark. The cells were
then visualized under a fluorescence microscope (BX50-FLA; Olympus
Corporation, Tokyo, Japan). Condensed, fractured or distorted
nuclei were detected in apoptotic cells, whereas a normal nuclear
size and uniform fluorescence were observed in viable cells. The
percentage of apoptotic cells was evaluated as follows: Number of
apoptotic cells/(numbers of apoptotic cells + numbers of viable
cells) × 100. Percentage of cell viability was calculated using the
optical density (OD), as follows: (OD treatment group/OD control
group) × 100.
Immunohistochemical staining
The cells were cultured in glass cover slips that
were placed in 6-well microtiter plates for 24 h and then washed
three times with PBS. Next, the cells were immediately fixed with
ice-cold 4% paraformaldehyde solution for 15 min, washed three
times with PBS, air-dried for 5 min and then incubated with 0.5%
Triton X-100 for 20 min. The cover slips were saturated with 5%
bovine serum albumin in PBS for 30 min at room temperature and then
processed for immunohistochemical staining with rabbit anti-Prx III
polyclonal primary antibody (ab73349; Abcam, Cambridge, MA, USA;
dilution, 1:400) for 4 h at 37°C. The primary antibodies were
removed by washing three times in PBS and the samples were
incubated for 1 h with goat anti-rabbit IgG horseradish
peroxidase-conjugated secondary antibodies (A0208; Beyotime
Institute of Biotechnology, Shanghai, China), prior to
visualization with diaminobenzidine for 10–15 min. Subsequently,
the cells were washed with distilled water and then counter stained
with hematoxylin (Beyotime Institute of Biotechnology).
Immunohistochemical micrograph was detected using a BX50-FLA
fluorescence microscope. Quantification of Prx III immunostaining
was performed by calculating the integral OD (IOD; positive area ×
average density) using an Image-Pro Plus system (Media Cybernetics,
Inc., Bethesda, MD, USA). The percentage of Prx III positive cells
was calculated as follows: (Number of Prx III positive cells/total
number of cells) × 100.
Western blot analysis
Cells were homogenized directly into cell lysis
buffer (Cell Signaling Technology, Inc., Danvers, MA, USA), and a
phosphatase inhibitor cocktail (Sigma-Aldrich), and the obtained
cell lysates were centrifuged at 12,000 × g for 10 min at 4°C.
Protein concentration was determined using a BCA protein assay kit
(Beyotime Institute of Biotechnology), following the manufacturer's
instructions. The extracted proteins were mixed with 5% sodium
dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis sample
buffer (Beyotime Institute of Biotechnology), then boiled at 100°C
for 7 min and separated by electrophoresis on a 10%
SDS-polyacrylamide gel. Subsequent to electrophoresis, the proteins
were transferred to polyvinylidene difluoride membranes, which were
then blocked in Tris-buffered saline-Tween 20 (TBS-T; 0.1% Tween
20) containing 5% non-fat dry milk, for 2 h at room temperature
with rotation. After blocking, the membranes were incubated with
the following antibodies: Rabbit anti-Prx III polyclonal antibody,
rabbit anti-FoxO3a polyclonal antibody (12829; Cell Signaling
Technologies, Inc.; dilution, 1:2,000), and rabbit
anti-phosphorylated-FoxO3a (anti-p-FoxO3a; Ser 253) polyclonal
antibody (13129; Cell Signaling Technologies, Inc.; dilution,
1:1,000). Subsequently, the membranes were incubated with 5% milk
or bovine serum albumin overnight at 4°C. The membranes were washed
three times in TBS-T to remove the primary antibody, and incubated
for 2 h with the appropriate horseradish peroxidase-conjugated
secondary antibodies. Following washing three times in TBS-T, the
antigen-antibody bands were detected using an enhanced
chemiluminescence reagent kit (Beyotime Institute of Biotechnology)
and quantified using Quantity One software (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). The data of the immunoblots of p-FoxO3a
were represented as a ratio of the phosphorylated forms to their
total forms.
Statistical analysis
Results are presented as the mean ± standard error
of mean. Statistical analysis was performed using Student's t-test
or analysis of variance with SPSS 13.0 software (SPSS, Inc.,
Chicago, IL, USA). In all cases, a value of P﹤0.05 was accepted as
indicating a statistically significant difference.
Results
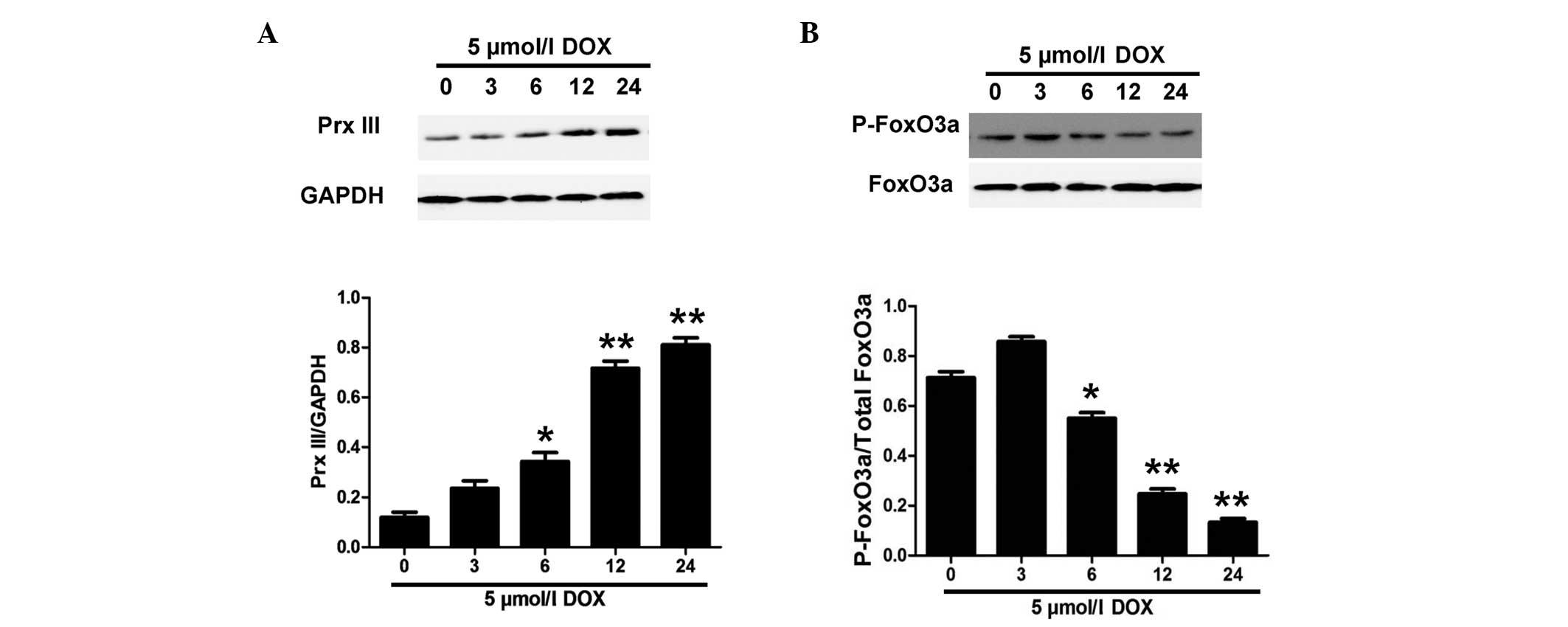
DOX increases Prx III expression in a
time-dependent manner
In order to elucidate whether Prx III was associated
with DOX-induced injuries in H9c2 cells, the expression of Prx III
was observed. H9c2 cells were treated with 5 µM DOX for the
indicated times (0, 3, 6, 12 and 24 h). Immunohistochemical
staining (Fig. 1) and western blot
analysis (Fig. 2A) revealed that Prx
III protein expression was significantly upregulated in H9c2 cells
after 6 h of incubation with 5 µM DOX. The expression of Prx III
increased as the incubation time was prolonged, with the strongest
effect observed in the 24 h group.
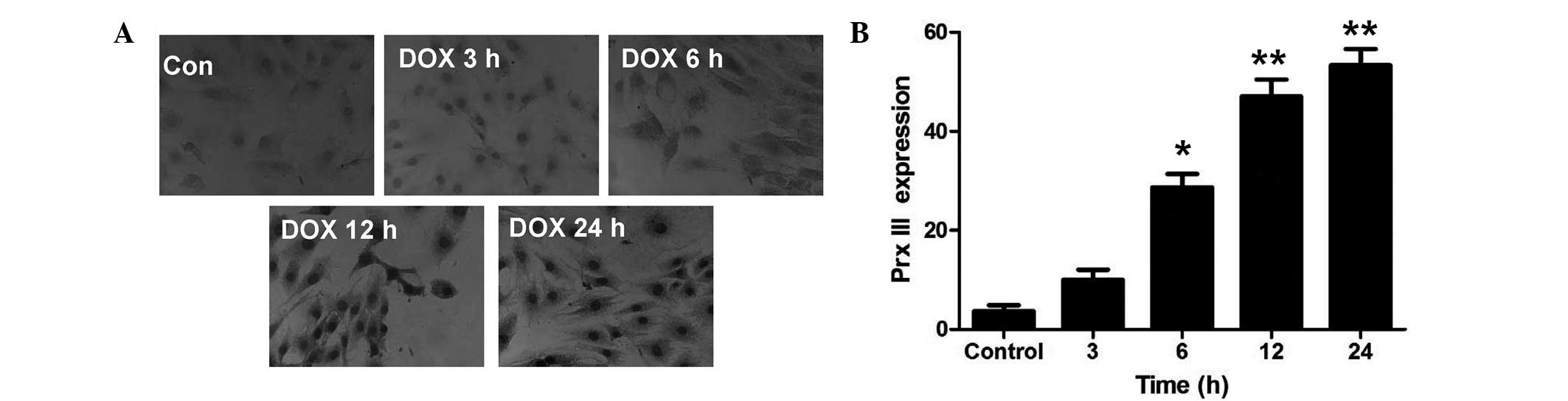 | Figure 1.Prx III expression in H9c2 cells
subsequent to DOX treatment for different durations. H9c2 cells
were treated with 5 µM DOX for 0 (control), 3, 6, 12 or 24 h.
Figure 1A: Immunohistochemical staining detected Prx III expression
in H9c2 cells under a fluorescence microscope. H9c2 cells were
treated with 5 µM DOX for 0, 3, 6, 12 or 24 h (magnification, x40).
Figure 1B: Quantitative analysis of the expression of Prx III in
H9c2 cells. Data are expressed as the mean ± standard error of mean
(n=3). *P<0.05 and **P<0.01 vs. control group. Prx III,
peroxiredoxin III; DOX, doxorubicin; Con, control. |
DOX treatment decreases p-FoxO3a
expression in H9c2 cells
A previous study reported that FoxO3a mediated Prx
III expression (13). The
aforementioned results (Figs. 1 and
2A) demonstrated that DOX treatment
induced Prx III expression in H9c2 cells; subsequently, we
attempted to investigate whether FoxO3a regulates the expression of
Prx III in DOX-induced H9c2 cell injury. Therefore, the expression
of p-FoxO3a was evaluated in the present study. H9c2 cells were
treated with 5 µM DOX for the indicated times (0, 3, 6, 12 and 24
h). As expected, DOX treatment significantly decreased the
expression of p-FoxO3a in a time-dependent manner (Fig. 2B). However, 5 µM DOX alone did not
induce significant changes in the expression of total FoxO3a. These
findings suggest that the expression of non-phosphorylated FoxO3a
was significantly increased following DOX stimulation, which
induced Prx III expression in H9c2 cells (Fig. 2A).
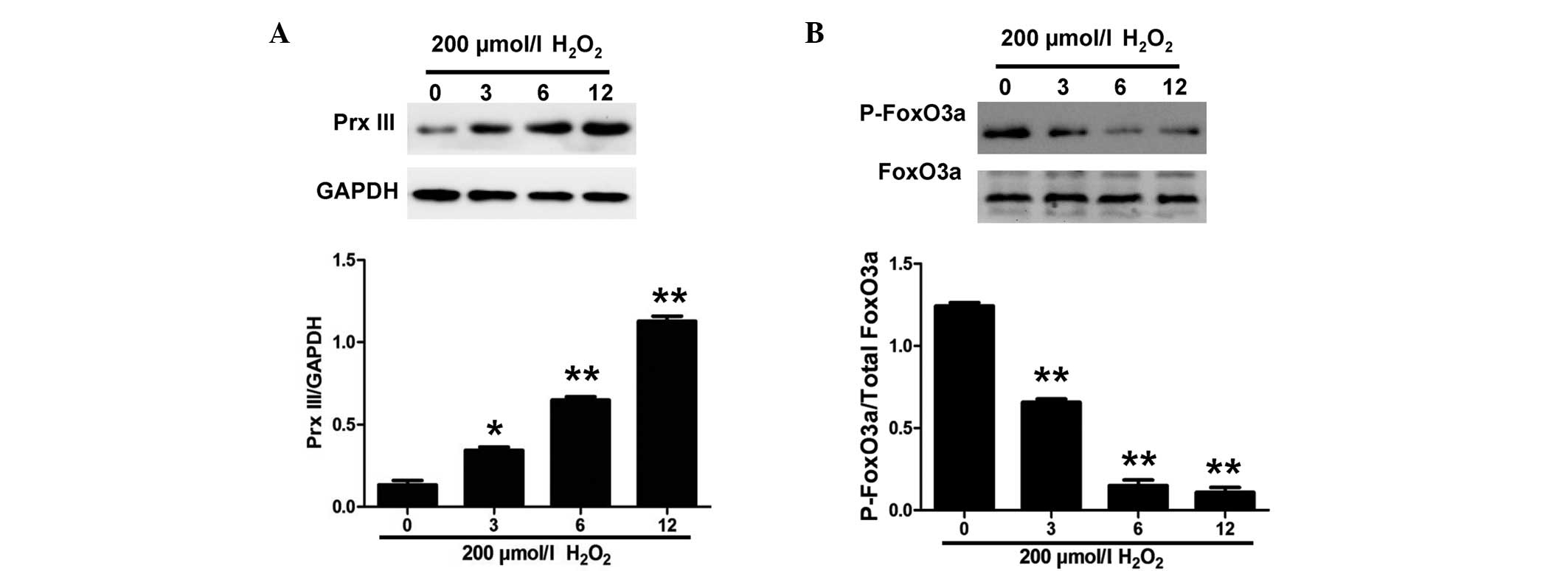
FoxO3a is required for the expression
of Prx III in H9c2 cells
In order to determine whether oxidative stress was
involved in the induction of Prx III expression by FoxO3a,
exogenous H2O2 was used to induce oxidative
stress in the H9c2 cells (Fig. 3).
As shown in Fig. 3B, the exposure of
H9c2 cells to 200 µmol/l H2O2 for the
indicated times (0, 3, 6 and 12 h) caused a significant
downregulation of p-FoxO3a expression in H9c2 cells. In addition,
the expression of Prx III following H2O2
treatment was examined and was found to increase (Fig. 3A). These data suggested that
oxidative stress induces Prx III expression in a FoxO3a-dependent
manner in H9c2 cells.
Oxidative stress on the expression of
Prx III and p-FoxO3a in H9c2 cells
To further confirm whether the DOX-induced
expression of Prx III is associated with oxidative stress, H9c2
cells were pretreated with 1,000 µM NAC (a ROS scavenger) for 60
min prior to exposure to 5 µM DOX for 24 h. As shown in Fig. 4, the pretreatment of cells with NAC
for 60 min markedly increased the expression of p-FoxO3a and
depressed the expression of Prx III. However, treatment with 1,000
µM NAC alone did not significantly alter the expression of total
FoxO3a. The results revealed that oxidative stress contributed to
the DOX-induced Prx III expression in a FoxO3a-dependent
manner.
Oxidative stress mediated DOX-induced
cytotoxicity and apoptosis in H9c2 cells
As shown in Fig. 5,
exposure of H9c2 cells to DOX at 5 µM for 24 h induced marked
cytotoxicity, leading to a decrease in cell viability. As shown in
Fig. 6, H9c2 cells treated with 5 µM
DOX for 24 h exhibited typical characteristics of apoptosis,
including condensation of chromatin, shrinkage of nuclei and
apoptotic bodies. To elucidate whether oxidative stress involved in
DOX-induced cytotoxicity and apoptosis, H9c2 cells were
preconditioned with a well-known ROS scavenger, NAC (1,000 µM)
prior to DOX treatment. The results showed that pretreatment of
cells with NAC significantly attenuated DOX-induced cytotoxicity
(Fig. 5) and apoptosis (Fig. 6). In addition, the exogenous ROS
H2O2 induced marked cytotoxicity and
apoptosis, exhibiting a similar function to that of DOX. These
results indicate that oxidative stress contributes to DOX-induced
cytotoxicity and apoptosis in H9c2 cells.
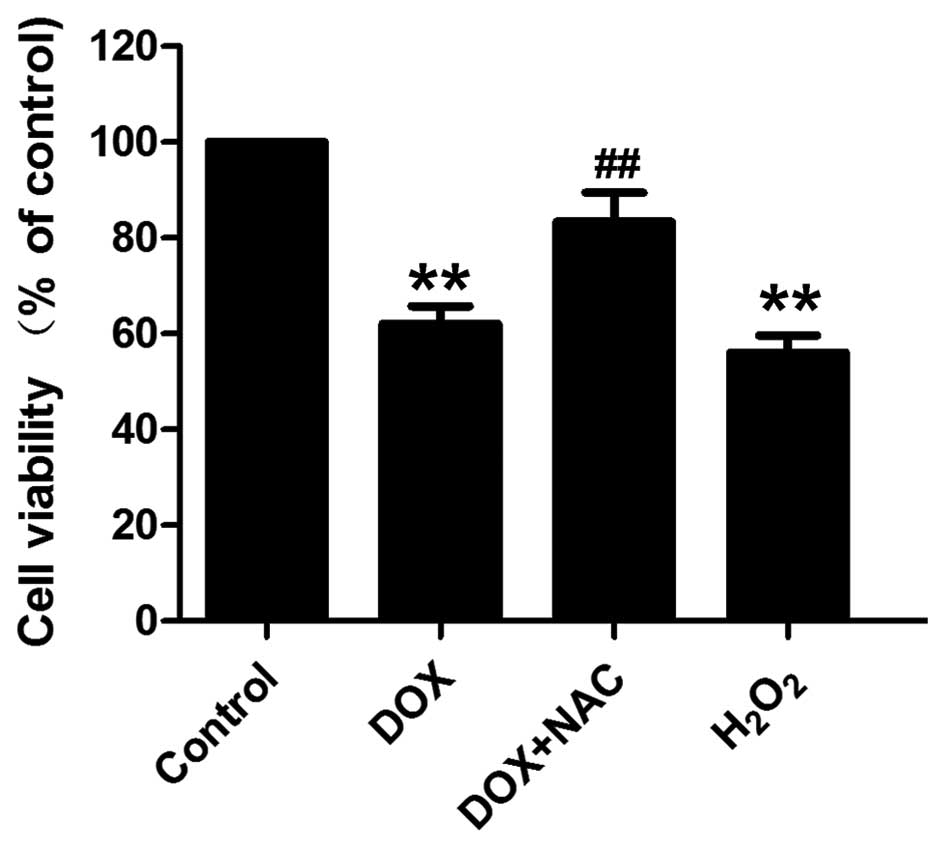 | Figure 5.Cell viability was measured using an
MTT assay, in order to determine the effect of antioxidant on
DOX-induced cytotoxicity in H9c2 cells. Data are expressed as the
mean ± standard error of mean (n=3). **P<0.01, vs. control
group; ##P<0.01, vs. DOX-treated group. DOX,
doxorubicin; NAC, N-acetyl-L-cysteine; control group, untreated
H9c2 cells; DOX group, cells treated with 5 µM DOX for 24 h; DOX +
NAC group, cells treated with 1,000 µM NAC for 60 min prior to
exposure to DOX; H2O2 group, cells treated
with 200 µM H2O2 for 12 h. |
 | Figure 6.Effect of antioxidant on DOX-induced
apoptosis in H9c2 cells. (A) Hoechst 33258 nuclear staining
followed by fluorescence imaging, performed to observe cell
apoptosis (magnification, x40). (B) The apoptotic rate was analyzed
with a cell counter and Image J 1.41o software. Data are expressed
as the mean ± standard error of mean (n=3). **P<0.01, vs.
control group; ##P<0.01, vs. DOX-treated group. DOX,
doxorubicin; NAC, N-acetyl-L-cysteine; control group, untreated
H9c2 cells; DOX group, cells treated with 5 µM DOX for 24 h; DOX +
NAC group, cells treated with 1,000 µM NAC for 60 min prior to
exposure to DOX; H2O2 group, cells treated
with 200 µM H2O2 for 12 h. |
Discussion
Doxorubicin (DOX) is one of the most widely used and
efficient antitumor drugs. However, its clinical use is limited by
its severe cumulative dose-associated cardiotoxicity (14). Numerous studies have demonstrated
that ROS generation due to the catalytic quinone moiety of DOX is
the major molecular mechanism involved in DOX-induced cardiac
toxicity, inducing cardiomyocyte apoptosis (15).
Prx III, a member of the Prx family, is a
mitochondrial antioxidant protein that is capable of catalyzing
H2O2 reduction (16). Prx III overexpression has been
reported to protect neurons against cell death induced by oxidative
stress (17). Due to these
characteristics, Prx III is an important candidate for the
treatment against left ventricular failure after myocardial
infarction, during which an increased production of ROS has been
observed within the mitochondria (11). Although various studies have
previously demonstrated the beneficial effects of antioxidants on
heart failure (18), no previous
studies have specifically investigated the protective role of Prx
III in DOX-induced cytotoxicity, to the best of our knowledge. In
the present study, Prx III was found to be significantly increased
in an injury model established by DOX-treatment in H9c2 cells.
Similar to the findings of the current study, an increase in Prx
III expression was previously reported by Xi et al (19), and nitrate treatment was found to
completely restore the expression of Prx III.
FoxO3a has been recently shown to be a key
transcription factor involved in resistance to oxidative stress
(20). When cells are exposed to
oxidative stress, FoxO3a translocates to the nucleus and activates
transcription by specifically binding to the consensus sequence
TTGTTTAC in the promoters of target genes (21). A previous study revealed that FoxO3a
increased the resistance to oxidative stress by upregulating the
expression of Prx III in human cardiac fibroblasts (13). Therefore, the present study evaluated
the regulation of FoxO3a in Prx III expression. Treatment of H9c2
cells with DOX was found to significantly inhibit the expression of
p-FoxO3a in a time-dependent manner. Following the downregulation
of p-FoxO3a expression, Prx III expression was significantly higher
in H9c2 cells treated with DOX, suggesting an indispensable role of
Prx III in the protection against oxidative stress.
Increasing evidence has suggested a major role for
ROS in the pathogenesis of cardiac failure (22). Furthermore, antioxidants have been
demonstrated to exert protective and beneficial effects against
heart failure (11). DOX induces
cardiomyocyte insult mainly by oxidative stress. In the current
study, in order to determine whether oxidative stress was involved
in the induction of Prx III expression by FoxO3a, exogenous
H2O2 was used to induce oxidative stress in
H9c2 cells. As evidenced in the present study,
H2O2 mimicked the effect of DOX, resulting in
a decrease in p-FoxO3a expression and an increase in Prx III
expression, after H9c2 cells were treated with
H2O2. The upregulation of Prx III in H9c2
cells would help cells to remove excessive ROS, providing a
favorable microenvironment for cell proliferation and enhancing
cardiomyocyte survival. These findings suggest that ROS may
function as an important mediator in the induction of Prx III
expression by FoxO3a following DOX treatment in H9c2 cells.
To further confirm that FoxO3a plays an essential
role in the mediation of Prx III expression in a DOX-treated H9c2
cell injury model, H9c2 cells were treated with 1,000 µmol/l NAC (a
ROS scavenger) for 60 min prior to exposure to DOX. The results
indicated that NAC significantly increased p-FoxO3a expression,
resulting in the suppression of Prx III expression. The results
also revealed that the antioxidant effect of NAC suppressed the
DOX-induced Prx III expression, suggesting that the expression of
Prx III was dependent on FoxO3a. These findings suggest that FoxO3a
regulated the expression of Prx III and protected against oxidative
stress by increasing Prx III expression.
Peroxiredoxin (Prx) III is an antioxidant enzyme
that controls cytokine-induced peroxide levels. In a previous
study, Jeong et al (9)
suggested that FoxO3a mediates the neuronal levels of the
expression of Prx III and the levels of expression of Mn-SOD in
vivo. The present results are consistent with these previous
findings, demonstrating for the first time that mitochondrial Prx
III was upregulated in DOX-treated H9c2 rat embryonic
cardiomyocytes. Jeong et al (9) and the present study demonstrated that
oxidative stress altered the expression of Prx III, suggesting that
Prx III may be used as a novel therapeutic targeting DOX-induced
cytotoxicity.
In conclusion, the present study demonstrated for
the first time that mitochondrial Prx III was upregulated in
DOX-treated H9c2 rat embryonic cardiomyocytes. The study provided
evidence that Prx III is an important regulator of intracellular
ROS, suggesting that upregulation of Prx III expression may be used
as a novel therapeutic strategy to protect against DOX-induced
cardiotoxicity.
Acknowledgements
This study was supported by grants from the Medical
Scientific Research Funds of Guangdong province (no. A2014810) and
the Graduate Student Research Innovation Project of Hunan province
(no. CX2013B397).
References
|
1
|
Magnano LC, Martínez Cibrian N, Andrade
González X and Bosch X: Cardiac complications of chemotherapy: Role
of prevention. Curr Treat Options Cardiovasc Med. 16:3122014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jang WJ, Choi DY and Jeon IS: Vascular
endothelial dysfunction after anthracyclines treatment in children
with acute lymphoblastic leukemia. Korean J Pediatr. 56:130–134.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Truong J, Yan AT, Cramarossa G and Chan
KK: Chemotherapy-induced cardiotoxicity: detection, prevention and
management. Can J Cardiol. 30:869–878. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Spagnuolo RD, Recalcati S, Tacchini L and
Cairo G: Role of hypoxia-inducible factors in the
dexrazoxane-mediated protection of cardiomyocytes from
doxorubicin-induced toxicity. Br J Pharmacol. 163:299–312. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Klotz LO, Sánchez-Ramos C, Prieto-Arroyo
I, Urbánek P, Steinbrenner H and Monsalve M: Redox regulation of
FoxO transcription factors. Redox Biol. 6:51–72. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu MH, Yuan C, He J, Tan TP, Wu SJ, Fu
HY, Liu J, Yu S, Chen YD, Le QF, et al: Resveratrol protects PC12
cells from high Glucose-induced neurotoxicity via PI3K/Akt/FoxO3a
pathway. Cell Mol Neurobiol. 35:513–522. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fiuza B, Subelzú N, Calcerrada P,
Straliotto MR, Piacenza L, Cassina A, Rocha JB, Radi R, de Bem AF
and Peluffo G: Impact of SIN-1-derived peroxynitrite flux on
endothelial cell redox homeostasis and bioenergetics: Protective
role of diphenyl diselenide via induction of peroxiredoxins. Free
Radic Res. 49:122–132. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Poynton RA and Hampton MB: Peroxiredoxins
as biomarkers of oxidative stress. Biochim Biophys Acta.
1840:906–912. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jeong HJ, Jeong HW, Song SS, Kang JW, Seo
JH, Lee YH, Lee KS and Kim DW: Upregulation of peroxiredeoxin III
in the hippocampus of acute immobilization stress model rats and
the Foxo3a-dependent expression in PC12 cells. Cell Mol Neurobiol.
31:1041–1046. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chae HZ, Kim HJ, Kang SW and Rhee SG:
Characterization of three isoforms of mammalian peroxiredoxin that
reduce peroxides in the presence of thioredoxin. Diabetes Res Clin
Pract. 45:101–112. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Matsushima S, Ide T, Yamato M, Matsusaka
H, Hattori F, Ikeuchi M, Kubota T, Sunagawa K, Hasegawa Y, Kurihara
T, et al: Overexpression of mitochondrial peroxiredoxin-3 prevents
left ventricular remodeling and failure after myocardial infarction
in mice. Circulation. 113:1779–1786. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chang TS, Cho CS, Park S, Yu S, Kang SW
and Rhee SG: Peroxiredoxin III, a mitochondrion-specific
peroxidase, regulates apoptotic signaling by mitochondria. J Biol
Chem. 279:41975–41984. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chiribau CB, Cheng L, Cucoranu IC, Yu YS,
Clempus RE and Sorescu D: FOXO3A regulates peroxiredoxin III
expression in human cardiac fibroblasts. J Biol Chem.
283:8211–8217. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lipshultz SE, Karnik R, Sambatakos P,
Franco VI, Ross SW and Miller TL: Anthracycline-related
cardiotoxicity in childhood cancer survivors. Curr Opin Cardiol.
29:103–112. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Salazar-Mendiguchía J, González-Costello
J, Roca J, Ariza-Sole A, Manito N and Cequier A:
Anthracycline-mediated cardiomyopathy: Basic molecular knowledge
for the cardiologist. Arch Cardiol Mex. 84:218–223. 2014.PubMed/NCBI
|
|
16
|
Song IS, Kim HK, Jeong SH, Lee SR, Kim N,
Rhee BD, Ko KS and Han J: Mitochondrial peroxiredoxin III is a
potential target for cancer therapy. Int J Mol Sci. 12:7163–7185.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hattori F, Murayama N, Noshita T and
Oikawa S: Mitochondrial peroxiredoxin-3 protects hippocampal
neurons from excitotoxic injury in vivo. J Neurochem. 86:860–868.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Betge S, Lutz K, Roskos M and Figulla HR:
Oral treatment with probucol in a pharmacological dose has no
beneficial effects on mortality in chronic ischemic heart failure
after large myocardial infarction in rats. Eur J Pharmacol.
558:119–127. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xi L, Zhu SG, Hobbs DC and Kukreja RC:
Identification of protein targets underlying dietary
nitrate-induced protection against doxorubicin cardiotoxicity. J
Cell Mol Med. 15:2512–2524. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Storz P: Forkhead homeobox type O
transcription factors in the responses to oxidative stress.
Antioxid Redox Signal. 14:593–605. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pierrou S, Hellqvist M, Samuelsson L,
Enerbäck S and Carlsson P: Cloning and characterization of seven
human forkhead proteins: binding site specificity and DNA bending.
EMBO J. 13:5002–5012. 1994.PubMed/NCBI
|
|
22
|
Schwarzer M, Osterholt M, Lunkenbein A,
Schrepper A, Amorim P and Doenst T: Mitochondrial reactive oxygen
species production and respiratory complex activity in rats with
pressure overload-induced heart failure. J Physiol. 592:3767–3782.
2014. View Article : Google Scholar : PubMed/NCBI
|