Introduction
The completion of the Human Genome Project genomic
map has led to significant advances in the field of gene therapy,
one being the development of RNA interference (RNAi). To implement
RNAi, the specific small interfering RNA (siRNA) for the target
gene must firstly be designed and synthesized; the oligonucleotide
sequence can then be designed and synthesized according to its
sequence in order to construct a recombinant lentiviral
plasmid-expressing short hairpin RNA (shRNA) (1,2). The
selection of the viral vector for the implementation of RNAi is
also important. Certain viral vectors used for gene transfer are
nonintegrative, whereas others are able to integrate into the host
cell chromatin. The most commonly used nonintegrative vectors are
derived from adenoviruses, herpes virus or adeno-associated virus.
The integrative vectors are derived from type C retroviruses, such
as murine leukemia virus or lentiviruses such as human
immunodeficiency virus. Lentiviral vectors are a widely used type
of virus vector for studies of gene function and therapy. These
vectors can be integrated into the genome of target cells and are
associated with a long expression duration, low immunogenicity and
low cytotoxicity, meaning that they can be effectively used in cell
lines that are considered to be difficult to transfect, including
macrophages, neurons and pancreatic cells (3–8). Tumor
necrosis factor-α (TNF-α) is an important proinflammatory cytokine
associated with inflammatory arthritis (9) that is involved in the
physiopathological process of rheumatoid arthritis. The aim of the
present study, therefore, was to construct an RNAi lentiviral
vector targeting the mouse TNF-α gene, in order to provide a basis
for RNAi gene therapy targeting TNF-α.
Materials and methods
Design and synthesis of siRNA
Based on the mouse TNF-α gene mRNA sequence
(NM_013693) in the GenBank database (http://www.ncbi.nlm.nih.gov/genbank/), siRNA was
designed according to siRNA design principles (10) and the siRNA Target Finder found on
the Ambion® (USA) website (https://www.lifetechnologies.com/cn/zh/home/life-science/rnai/synthetic-rnai-analysis.html#tool).
In order to avoid off-target effects (11), the Basic Local Alignment Search Tool
(http://blast.ncbi.nlm.nih.gov/Blast.cgi) was run
against the siRNA sequence so that homologous sequences could be
excluded; the negative control was designed according to the
literature (12–14). The detailed sequences are shown in
Table I. siRNA was synthesized by
Zimmer (Shanghai) Medical International Trading Co., Ltd.
(Shanghai, China).
 | Table I.siRNA sequences. |
Table I.
siRNA sequences.
| Name | Sequence |
|---|
| siRNA1 | Sense:
5′-CAGAAAGCAUGAUCCGCGATT-3′ |
|
| Antisense:
5′-UCGCGGAUCAUGCUUUCUGTG-3′ |
| siRNA2 | Sense:
5′-GUGCCUAUGUCUCAGCCUCUUTT-3′ |
|
| Antisense:
5′-AAGAGGCUGAGACAUAGGCACTT-3′ |
| siRNA3 | Sense:
5′-GCCGAUUUGCUAUCUCAUATT-3′ |
|
| Antisense:
5′-UAUGAGAUAGCAAAUCGGCTG-3′ |
| Negative control
siRNA | Sense:
5′-UUCUCCGAACGUGUCACGUTT-3′ |
Cell culture and transfection
RAW264.7 mouse macrophage cells (Shanghai Cell Bank
of China Academy of Medical Sciences, Shanghai, China) were
cultured in Dulbecco's modified Eagle's medium (DMEM) containing
10% fetal bovine serum (Gibco; Life Technologies, Carlsbad, CA,
USA) in a 37°C incubator with 5% CO2. The RAW264.7 cells
were inoculated in 24-well cell culture plates at a density of
8×104/well and underwent transfection at 40% confluence.
The final concentration of siRNA was 100 nM (15). The transfection was performed using
X-tremeGENE™ siRNA Transfection Reagent (Roche Applied Science,
Indianapolis, IN, USA), according to the manufacturer's
instructions. Three control groups were established: A blank
control group, with an equal volume of medium; a transfection
reagent control group and a negative control group. Each group was
set-up in three repeated wells. A total of 5 h after the completion
of the siRNA transfection, lipopolysaccharide (LPS; Sigma-Aldrich,
St. Louis, MO, USA) was added at a final concentration of 1 µg/µl
to each well. Following 2 h of incubation, the cells and
supernatant were collected for use in ELISA and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
detection. The experiments were repeated three times.
RT-qPCR
Total RNA was extracted from the cells in each group
using the Takara RNA extraction kit, in accordance with the
manufacturer's instructions (Takara Biotechnology Co., Ltd.,
Dalian, China). Following extraction, 1 µg total RNA was subjected
to RT to obtain cDNA for qPCR analysis. The mouse TNF-α,
interleukin (IL)-1β, IL-6, β-actin primers and Taqman probe were
designed using an application of the Primer Express™ software v2.0
(Applied Biosystems, Foster City, CA, USA), and the sequences were
as shown in Table II. The primers
were synthesized by Shanghai Sangon Biotechnology Co., Ltd.
(Shanghai, China), and the probes were synthesized by Takara
Biotechnology Co., Ltd. The RT-qPCR system and reaction conditions
were implemented based on the instructions of the PrimeScript™
RT-PCR kit (Takara Biotechnology Co., Ltd.). The qPCR was performed
in the Rotor-Gene RG3500 PCR reaction instrument (Qiagen, Shanghai,
China). The Ct value was automatically recorded by Rotor-Gene6
software. The relative expression levels of the TNF-α, IL-1β and
IL-6 mRNA were calculated using the 2−ΔΔCt method: ΔΔCt
= (Ct target gene - Ct reference
gene)test group - (Ct target gene - Ct
reference gene)blank control group. The
inhibition rate of mRNA expression in each group was calculated
using the following formula: mRNA inhibition rate = [(1-relative
expression level of mRNAtreatment group/mRNA relative
expressionnegative control group)/1] × 100%.
| Table II.Sequences of the primers and
probes. |
Table II.
Sequences of the primers and
probes.
| Gene | Primers and
probes | Annealing temperature
(°C) |
|---|
| TNF-α | Forward primer:
5′-TCTTCCCTGAGGTGCAATGC-3′ | 62 |
|
| Reverse primer:
5′-GCTCCGTTTTCACAGAAAACATG-3′ |
|
|
| Probe: 5′-(FAM)
TGGAGGACCCAGTGTGGGAAGCTGT(TAMRA)-3′ |
|
| IL-1β | Forward primer:
5′-GGAGCTCCCTTTTCGTGAATG-3′ | 62 |
|
| Reverse primer:
5′-AGGTAAGTGGTTGCCCATCAGA-3′ |
|
|
| Probe: 5′-(FAM)
CCAAGACAGGTCGCTCAGGGTCACA(TAMRA)-3′ |
|
| IL-6 | Forward primer:
5′-TCCTACCCCAATTTCCAATGC-3′ | 62 |
|
| Reverse primer:
5′-CCACAGTGAGGAATGTCCACAA-3′ |
|
|
| Probe: 5′-(FAM)
ATCTACTCGGCAAACCTAGTGCGTT(TAMRA)-3′ |
|
| β-actin | Forward primer:
5′-ATGGTGGGAATGGGTCAGAAG-3′ | 62 |
|
| Reverse primer:
5′-TCCATGTCGTCCCAGTTGGTA-3′ |
|
|
| Probe: 5′-(FAM)
TGACGAGGCCCAGAGCAAGAGAGGT(TAMRA)-3′ |
|
ELISA
The supernatant of the cell culture in each group
was used to detect the TNF-α content via the ELISA method,
according to the kit manufacturer's instructions (Wuhan Boster
Biological Technology, Ltd., Wuhan, China). The protein inhibition
rate of TNF-α of each group was calculated using the following
formula: Protein inhibition rate = [(TNF-αnegative control
group-TNF-αtreatment group)/TNF-αnegative
control group] × 100%.
Design of shRNA
Having screened for the high-efficiency siRNA
sequence, the oligonucleotide sequence was designed and synthesized
according to the requirements of the lentiviral vector
construction. The 21-nucleotide oligonucleotide sequence in the
forward single strand was forward and reverse combined, during
which the loop structure was added to make the oligonucleotide
forming the hairpin structure, with the restriction enzyme cutting
site at the two ends. The sequence of the duplex structure was as
follows: 5′-CCG GAA GAG GCT GAG ACA TAG GCA CTT CAA GAG AGT GCC TAT
GTC TCA GCC TCT TTT TTTG-3′; 5′-AAT TCA AAA AAA GAG GCT GAG ACA TAG
GCA CTC TCT TGA AGT GCC TAT GTC TCA GCC TCTT-3′.
Construction of recombinant lentivirus
shuttle plasmid
The double-stranded DNA was formed by
oligonucleotides annealing at the reaction conditions of 90°C for 4
min, 70°C for 10 min and a slow decrease to room temperature. The
PGCSIL-green fluorescent protein (GFP) vector (Shanghai Jikai
Industry, Co., Shanghai, China) was double enzyme digested by
AgeI and EcoRI at 37°C for 1 h, and gel
electrophoretic recovery of the linear pGCSIL-GFP fragments was
performed. The annealed, double-stranded DNA and linear pGCSIL-GFP
fragments were connected using T4 ligase (Takara Biotechnology Co.,
Ltd.) at 4°C for 16 h. The pGCSIL-GFP-shRNA connection products
were used to transform DH5α competent cells (Takara Biotechnology
Co., Ltd.), and the transformed competent cells were then shifted
into antibiotic LB medium containing ampicillin (Shanghai Solarbio
Bioscience & Technology Co., Ltd., Shanghai, China), at 37°C
for 16 h. A single colony was selected to undergo PCR. The PCR
primers were as follows: Upstream, 5′-CCTATTTCCCATGATTCCTTCATA-3′;
and downstream, 5′-GTAATACGGTTATCCACGCG-3′. The PCR reaction
conditions comprised initial denaturation at 94°C for 30 sec;
denaturation at 94°C for 30 sec, annealing at 60°C for 30 sec and
extension at 72°C for 30 sec (30 cycles); and then extension at
72°C for 6 min. The PCR products were subjected to 2% agarose gel
electrophoresis. Following the selection of the single colony and
the agitation of the bacteria, the plasmid was extracted for
sequencing.
Lentiviral packaging
293T cells (Type Culture Collection of the Chinese
Academy of Sciences, Shanghai, China) were cultured in DMEM
containing 10% fetal bovine serum in a 37°C incubator with 5%
CO2. One day before transfection, the logarithmic growth
phase cells were inoculated at a density of 6×108/l in
15-cm cell culture dishes (70–80% confluence at the time of
transfection). At 2 h before transfection, the cell medium was
changed to serum-free medium. The lentiviral shuttle plasmid,
lentiviral packaging plasmid and transfection reagent
Lipofectamine™ 2000 (Invitrogen; Life Technologies) were mixed to
form transfection complexes, which were co-transfected into 293T
cells. At 8 h after transfection, the medium was replaced with
fresh medium containing 10% serum and the cells were then cultured
for a further 48 h, prior to the culture supernatant being
collected, centrifuged at 4,000 × g for 10 min and filtered with a
0.45-µM filter. The concentrated vector, which was referred to as
lentivirus-shTNF, was stored at −80°C until use.
Determination of virus titer
The 293T cells that were used for virus transfection
were inoculated in 96-well plates at a density of
4×104/well and cultured in a 37°C incubator with 5%
CO2 for 24 h. A total of 90 µl culture medium was
removed and 90 µl serial dilution of virus stock was added, prior
to further culture for 24 h and the replacement of the medium.
After 96 h, the green fluorescence expression was observed under a
fluorescence microscope. The wells in which ~5 fluorescent cells as
well as the corresponding dilution ratio were recorded (fluorescent
cells were counted independently by 2 specifically assigned
individuals). The virus titer was calculated using the following
formula: Virus titer (TU/µl) = number of fluorescent
cells/corresponding dilution ratio.
Statistical analysis
The software SigmaStat 11.0 (Systat Software, Inc.,
San Jose, CA, USA) was used for data processing. Quantitative data
are expressed as the mean ± standard error of the mean; comparisons
among the groups were performed using one-way analysis of variance,
and the Dunnett's t-test was used for comparisons between
the experimental groups and the control group. P<0.05 was
considered to indicate a statistically significant difference.
Results
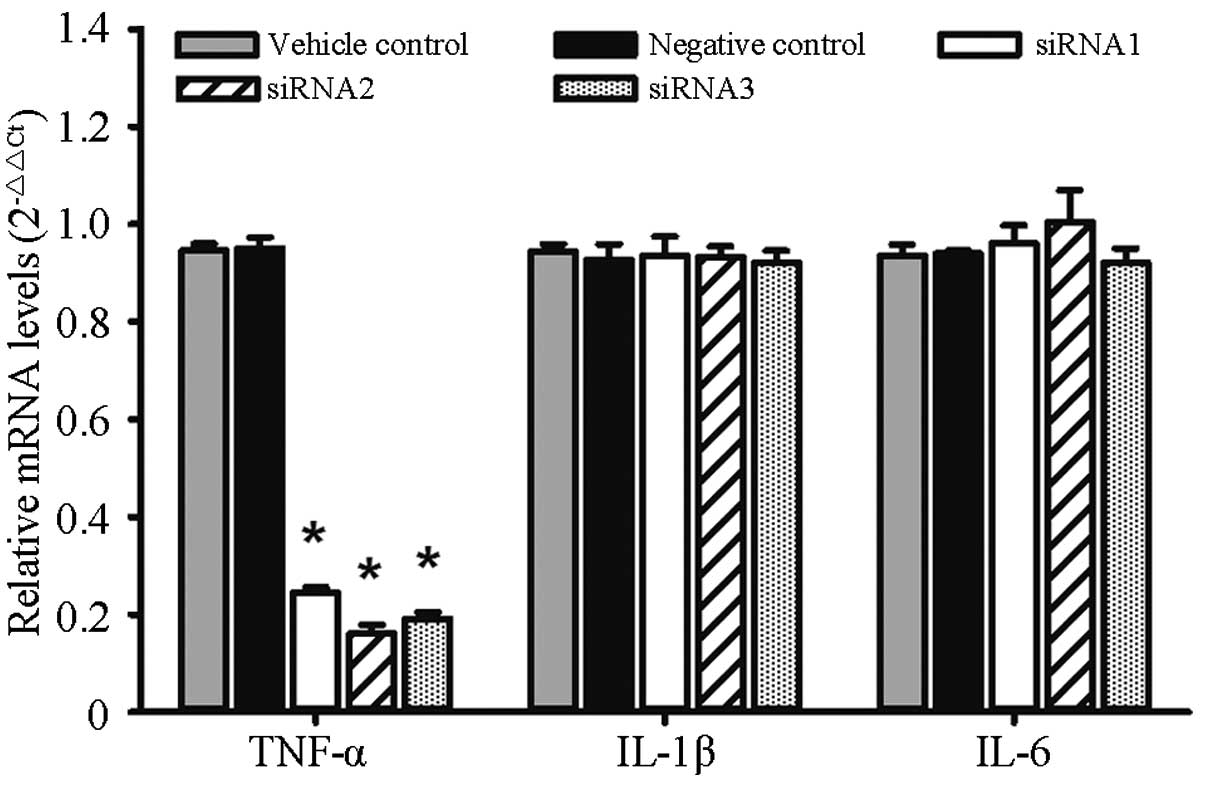
Expression of TNF-α, IL-1β and IL-6
mRNA
The relative mRNA expression levels of TNF-α in the
siRNA1, siRNA2, siRNA3 and negative control groups were,
respectively, 0.24±0.01, 0.16±0.02, 0.19±0.01 and 0.95±0.02.
Significant differences were found in the expression levels between
each group (F=531.3, P<0.0001). As compared with the negative
control group, the mRNA inhibition rates were 74.26, 83.09 and
79.93% for siRNA1, siRNA2 and siRNA3, respectively. The relative
mRNA expression levels of IL-1β in the siRNA1, siRNA2, siRNA3 and
negative control groups were, respectively, 0.93±0.04, 0.93±0.02,
0.92±0.02 and 0.93±0.03, and no significant differences were found
among the groups (F=0.981, P=0.9807). The relative mRNA expression
levels of IL-6 in the siRNA1, siRNA2, siRNA3 and negative control
groups were, respectively, 0.96±0.04, 1±0.07, 0.92±0.03 and
0.94±0.01, and no significant differences were found among the
groups (F=0.739, P=0.5867), as shown in Fig. 1.
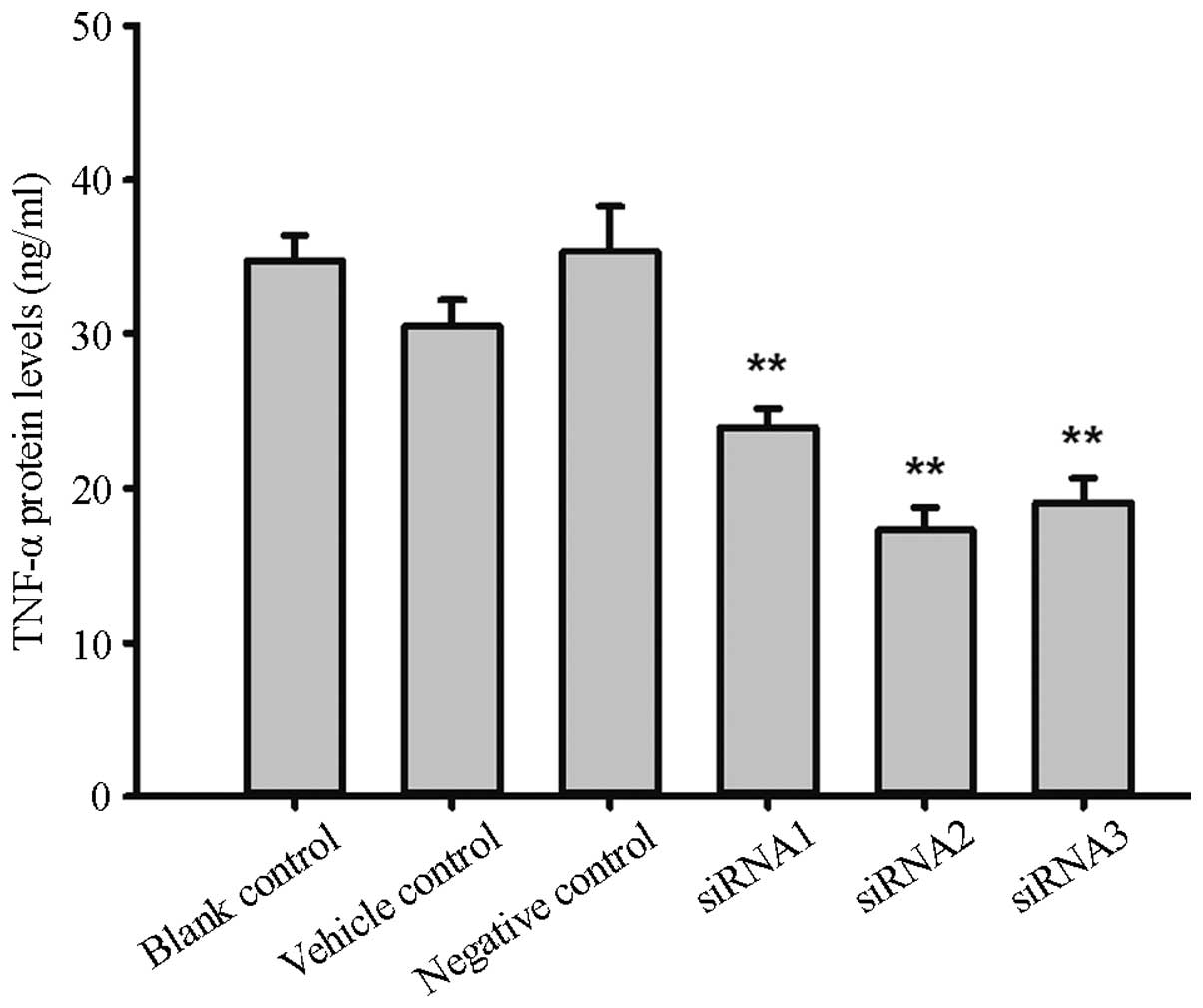
TNF-α protein expression
The protein expression levels of TNF-α in the
siRNA1, siRNA2, siRNA3 and negative control groups were 23.95±1.21,
17.27±1.46, 19.07±1.57 and 35.37±2.93 ng/ml respectively, and
significant differences were found between the groups (F=18.1,
P=0.0006). Compared with the negative control, the TNF-α protein
expression inhibition rates were 32.29, 51.16 and 46.08%,
respectively. The protein expression levels in the blank and
reagent control groups were 34.75±1.67 and 30.48±1.701 ng/ml,
respectively, and no significant difference was found compared with
the levels in the negative control group (F=1.49, P=0.30), as shown
in Fig. 2.
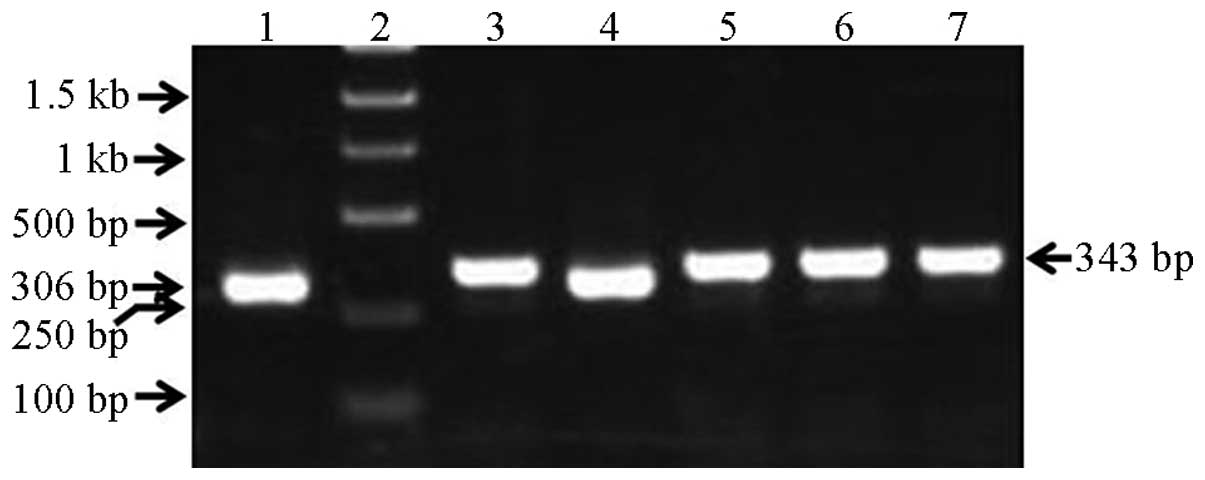
qPCR
The recombinant lentiviral plasmid PCR amplification
product was 343 bp, the empty-vector PCR product was 306 bp and the
insert fragment was 61 bp; these results were consistent with the
expectation. It should ne noted that there were 4 restriction
enzyme sites in the pGCSIL-GFP vector between the AgeI and
EcoRI sites, with a total length of 24 bp; therefore, the
products were as follows: 306 bp – 24 bp + insert fragment = 343
bp, as shown in Fig. 3.
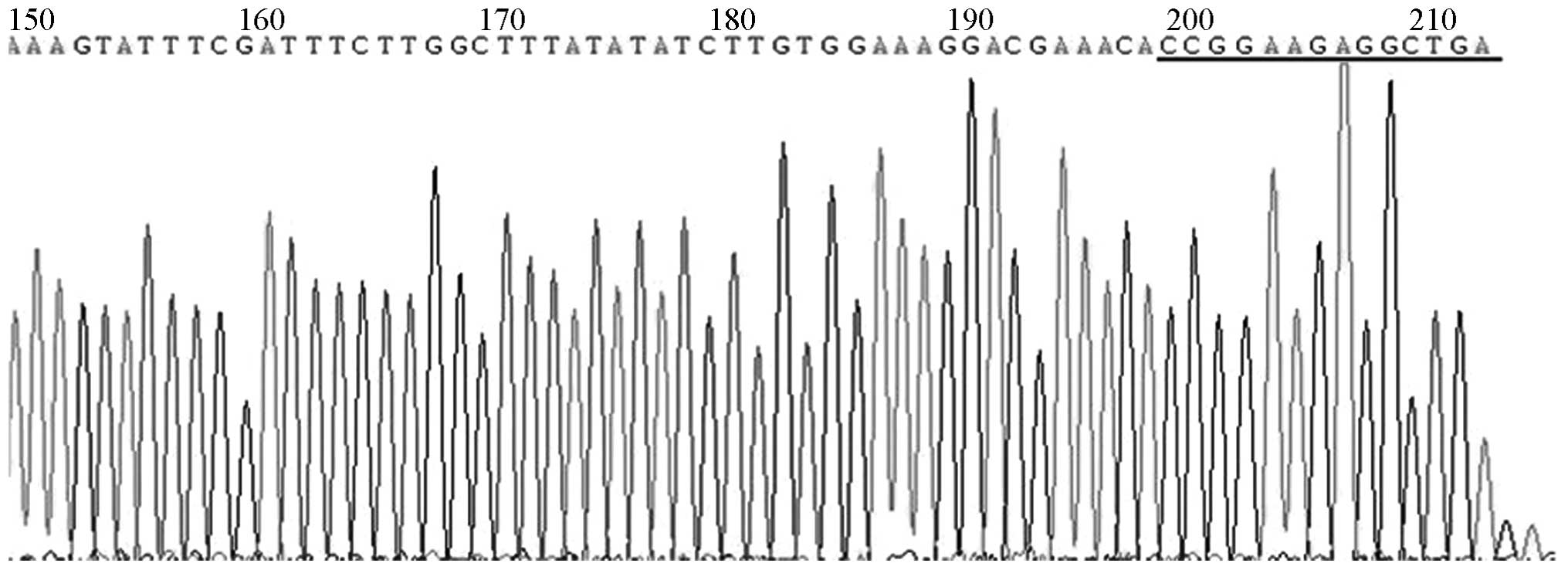
DNA sequencing
DNA sequencing revealed that the pGCSIL-GFP-shRNA
sequencing reaction was interrupted, but the presence of an
insertion sequence was indicated, as shown in Fig. 4.
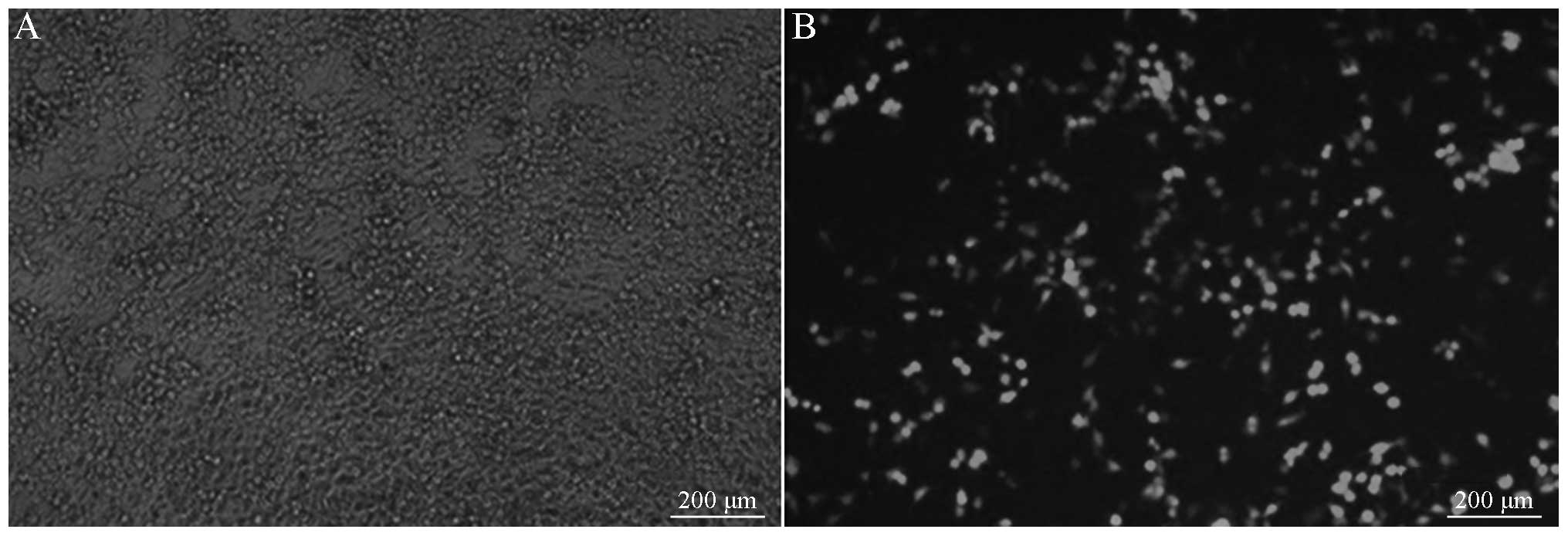
Lentiviral packaging and
identification
The lentiviral shuttle and packaging plasmids were
co-transfected into the 293T cells. After 48 h, it was visible that
cells were growing well, and the fluorescence expression was strong
under the fluorescence microscope. After 96 h, it was observed
under the fluorescence microscope that the number of fluorescent
cells decreased as the dilution ratio increased. The lentiviral
titer was calculated to be 2×106 TU/µl, as shown in
Fig. 5.
Discussion
TNF-α is the key cytokine leading to inflammatory
joint diseases (9); therefore, the
aim of the present study was to construct RNAi lentiviral vectors
targeting the mouse TNF-α gene, providing a basis for RNAi gene
therapy. Developing an efficient interference fragment and
effective gene transfer method is fundamental in RNAi gene
therapy.
In this study, three pairs of siRNA targeting TNF-α
were designed, synthesized and screened at the cellular level.
RAW264.7 mouse macrophage cells are commonly used as inflammatory
cell models, expressing high levels of TNF-α following stimulation
by LPS (16,17), and can thus be used for siRNA
screening. Following the transfection of the siRNA into the
RAW264.7 cells and the administration of the LPS stimulus, the RNAi
gene silencing effect was detected at the mRNA and protein levels.
The results showed that the expression inhibition of the three
siRNAs on TNF-α at the mRNA and protein levels was significantly
different compared with that in the control group; siRNA2 exhibited
the highest inhibitory effect at both investigated levels and was
selected as the highest silence efficiency interference sequence
for follow-up study.
The inhibition of TNF-α expression was expected be
accompanied by a decrease in the expression of IL-1β, IL-6 and
other inflammatory factors, as TNF-α can further induce the
generation of IL-1β and IL-6; however, the results showed that RNAi
targeting the TNF-α gene had no significant effect on the mRNA
expression of IL-1β or IL-6. This may have been a result of the
fact that, although the application of the siRNA inhibited the
expression of TNF-α, the inflammatory cell model was still in
existence due to the LPS stimulation, and the combination of LPS
and the RAW264.7 cell surface receptor was still able to induce the
generation of IL-1β and IL-6 via the activation of nuclear
factor-κB. The results therefore showed the specificity of the
RNAi.
In the present study, the siRNA was transfected into
the RAW264.7 cells, which were then stimulated by LPS; 7 h after
transfection, the gene silencing effect was observed. A study
involving the J774.1 mouse macrophage cell line described similar
results to those in the present study (15); however, the study reported that the
gene silencing effect of siRNA was observed at the mRNA level
>24 h after transfection and at the protein level >48 h after
transfection.
Liposomes and electrotransfection are common methods
of gene transfection; however, the target gene fragments following
transfection are easily degraded and, following
electrotransfection, cells have a low survival rate and low
availability in clinical experiments (18,19).
Viral vectors, such as adenovirus, adeno-associated virus and
lentiviral vectors, have been widely used in molecular biology;
however, adenoviruses are difficult to transfect into monocytes
(20). It has previously been shown
that lentiviral vectors can be used in numerous types of cells that
are difficult to transfect, including monocytes (6–8,21). In addition, lentiviral vectors can
result in the integration of the interference fragment of the
target gene into the genome, enabling the long-term suppression of
target gene expression. This is particularly advantageous for in
vitro and in vivo experimental studies and, therefore, a
lentiviral vector was selected for use in the RNAi in the present
study.
In this study, the pGCSIL-GFP vector containing a U6
promoter was used as the interference plasmid, which enabled the
sustained expression of shRNA in the host cells, as well as the
concurrent expression of GFP. In the identification of the
recombinant lentiviral shuttle plasmid PCR product, electrophoresis
showed that a 61-bp target fragment was inserted in the positive
clones. DNA sequencing only showed a partial insertion sequence,
which may have been due to the readiness of shRNA to form a
secondary structure (22,23). PCR product electrophoresis and DNA
sequencing determined that the recombination was successful. In
addition to the pGCSIL-GFP vector, the recombinant lentivirus
packaging system must contain a packaging vector with the other
elements and capsid protein for lentiviral packaging. The use of
293T cells as the virus packaging cells can produce high-titer
virus particles, which can be used to detect the virus packaging
effect and infection efficiency of target cells. The virus titer in
the present study was found to be 2×106 TU/µl,
indicating usefulness for subsequent in vitro and in
vivo experimental studies.
In conclusion, an RNAi lentiviral vector targeting
the TNF-α gene was in constructed the present study; the vector
construction was found to be successful and, following packaging
and concentration, high-titer lentiviral vectors were obtained. The
findings of this study provide a basis for subsequent experimental
study of RNAi gene therapy targeting TNF-α.
Acknowledgements
This study was supported by the Scientific Research
Award Fund of Shandong Province Outstanding Young Scientist.
References
|
1
|
Chen SY, Shiau AL, Li YT, Lin YS, Lee CH,
Wu CL and Wang CR: Suppression of collagen-induced arthritis by
intra-articular lentiviral vector-mediated delivery of Toll-like
receptor 7 short hairpin RNA gene. Gene Ther. 19:752–760. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sakuma T, Barry MA and Ikeda Y: Lentiviral
vectors: Basic to translational. Biochem J. 443:603–618. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gouze E, Pawliuk R, Pilapil C, Gouze JN,
Fleet C, Palmer GD, Evans CH, Leboulch P and Ghivizzani SC: In vivo
gene delivery to synovium by lentiviral vectors. Mol Ther.
5:397–404. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lewis PF and Emerman M: Passage through
mitosis is required for oncoretroviruses but not for the human
immunodeficiency virus. J Virol. 68:510–516. 1994.PubMed/NCBI
|
|
5
|
Naldini L, Blömer U, Gallay P, Ory D,
Mulligan R, Gage FH, Verma IM and Trono D: In vivo gene delivery
and stable transduction of nondividing cells by a lentiviral
vector. Science. 272:263–267. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wilson AA, Kwok LW, Porter EL, Payne JG,
McElroy GS, Ohle SJ, Greenhill SR, Blahna MT, Yamamoto K, Jean JC,
et al: Lentiviral delivery of RNAi for in vivo lineage-specific
modulation of gene expression in mouse lung macrophages. Mol Ther.
21:825–833. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wollebo HS, Woldemichaele B and White MK:
Lentiviral transduction of neuronal cells. Methods Mol Biol.
1078:141–146. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Houbracken I, Baeyens L, Ravassard P,
Heimberg H and Bouwens L: Gene delivery to pancreatic exocrine
cells in vivo and in vitro. BMC Biotechnol. 12:742012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Moelants EA, Mortier A, Van Damme J and
Proost P: Regulation of TNF-α with a focus on rheumatoid arthritis.
Immunol Cell Biol. 91:393–401. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Elbashir SM, Harborth J, Lendeckel W,
Yalcin A, Weber K and Tuschl T: Duplexes of 21-nucleotide RNAs
mediate RNA interference in cultured mammalian cells. Nature.
411:494–498. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Svoboda P: Off-targeting and other
non-specific effects of RNAi experiments in mammalian cells. Curr
Opin Mol Ther. 9:248–257. 2007.PubMed/NCBI
|
|
12
|
Lin X, Yu Y, Zhao H, Zhang Y, Manela J and
Tonetti DA: Overexpression of PKCalpha is required to impart
estradiol inhibition and tamoxifen-resistance in a T47D human
breast cancer tumor model. Carcinogenesis. 27:1538–1546. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Qiu Z, Huang C, Sun J, Qiu W, Zhang J, Li
H, Jiang T, Huang K and Cao J: RNA interference-mediated signal
transducers and activators of transcription 3 gene silencing
inhibits invasion and metastasis of human pancreatic cancer cells.
Cancer Sci. 98:1099–1106. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ai Z, Yin L, Zhou X, Zhu Y, Zhu D, Yu Y
and Feng Y: Inhibition of survivin reduces cell proliferation and
induces apoptosis in human endometrial cancer. Cancer. 107:746–756.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Khoury M, Louis-Plence P, Escriou V, Noel
D, Largeau C, Cantos C, Scherman D, Jorgensen C and Apparailly F:
Efficient new cationic liposome formulation for systemic delivery
of small interfering RNA silencing tumor necrosis factor alpha in
experimental arthritis. Arthritis Rheum. 54:1867–1877. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhu ZG, Jin H, Yu PJ, Tian YX, Zhang JJ
and Wu SG: Mollugin inhibits the inflammatory response in
lipopolysaccharide-stimulated RAW264.7 macrophages by blocking the
Janus kinase-signal transducers and activators of transcription
signaling pathway. Biol Pharm Bull. 36:399–406. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qureshi AA, Guan XQ, Reis JC, Papasian CJ,
Jabre S, Morrison DC and Qureshi N: Inhibition of nitric oxide and
inflammatory cytokines in LPS-stimulated murine macrophages by
resveratrol, a potent proteasome inhibitor. Lipids Health Dis.
11:762012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kusumawati A, Commes T, Liautard JP and
Widada JS: Transfection of myelomonocytic cell lines: Cellular
response to a lipid-based reagent and electroporation. Anal
Biochem. 269:219–221. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liao HS, Kodama T, Doi T, Emi M, Asaoka H,
Itakura H and Matsumoto A: Novel elements located at −504 to −399
bp of the promoter region regulated the expression of the human
macrophage scavenger receptor gene in murine macrophages. J Lipid
Res. 38:1433–1444. 1997.PubMed/NCBI
|
|
20
|
Wirtz S, Becker C, Blumberg R, Galle PR
and Neurath MF: Treatment of T cell-dependent experimental colitis
in SCID mice by local administration of an adenovirus expressing
IL-18 antisense mRNA. J Immunol. 168:411–420. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee JS, Hmama Z, Mui A and Reiner NE:
Stable gene silencing in human monocytic cell lines using
lentiviral-delivered small interference RNA. Silencing of the
p110alpha isoform of phosphoinositide 3-kinase reveals differential
regulation of adherence induced by
1alpha,25-dihydroxycholecalciferol and bacterial
lipopolysaccharide. J Biol Chem. 279:9379–9388. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guo Y, Liu J, Li YH, Song TB, Wu J, Zheng
CX and Xue CF: Effect of vector-expressed shRNAs on hTERT
expression. World J Gastroenterol. 11:2912–2915. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
McIntyre GJ and Fanning GC: Design and
cloning strategies for constructing shRNA expression vectors. BMC
Biotechnol. 6:12006. View Article : Google Scholar : PubMed/NCBI
|