Introduction
Pathological cardiac hypertrophy develops in
response to hemodynamic overload, which is a major predictor for
the development of coronary artery disease and heart failure
(1). Preventing and postponing the
progression of cardiac hypertrophy may serve as an effective
therapeutic strategy for the treatment of patients with heart
failure (2). Clinical management of
pathological cardiac hypertrophy is targeted against the underlying
cause of the condition and typically involves the administration of
certain pharmacological agents, such as
angiotensin-converting-enzyme inhibitor and angiotensin receptor
blockers (3). However, although
current pharmacological approaches appear to be beneficial in
improving the quality of life of patients with heart failure, they
do not markedly reduce the mortality rates (4). A previous study showed that puerarin
attenuates the cardiac hypertrophy induced by pressure overload by
downregulating phosphoinositide 3-kinase (PI3K)/protein kinase B
(Akt) and c-Jun N-terminal kinase (JNK) signaling pathways
(5). Therefore, a key future
challenge is the identification of pharmacological agents that
alleviate progressive myocardial dysfunction and unfavorable
remodeling, improve the quality of life and reduce the mortality
rate of patients with heart failure.
Naringenin is a bitter principle component of
grapefruit (Citrus paradisi) that exhibits a variety of
pharmacological effects, including anti-inflammatory (6), hypolipidemic, antithrombotic and
antiatherogenic properties (5). The
dietary consumption of citrus fruits is associated with a reduced
rate of acute coronary events (7),
and naringenin appears to exert beneficial effects on the
cardiovascular system (8).
Naringenin has been demonstrated to promote endothelium-independent
vasorelaxing effects on rat aortic rings that have been
precontracted with noradrenaline (9). Furthermore, naringenin may be able to
alleviate myocardial ischemia/reperfusion injury (10). In addition, due to its
radical-scavenging and iron-chelating properties, naringenin may
potentially protect against doxorubicin-induced cardiac toxicity
(6). However, it currently remains
clear whether naringenin is able to mitigate the development of
pressure overload-induced cardiac hypertrophy and postpone the
progression of heart failure.
In the present study, aortic banding (AB) was used
to induce cardiac hypertrophy in C57BL/6 mice in order to
investigate whether naringenin is able to exert a protective effect
against cardiac hypertrophy, fibrosis and left ventricular
dysfunction, and to identify the possible mechanisms underlying
these effects. In addition, the study aimed to determine whether
naringenin is able to influence the activation of mitogen-activated
protein kinase (MAPK) and PI3K/Akt signaling pathways, which are
activated by pressure overload.
Materials and methods
Animals and animal models
All the experimental procedures were performed in
accordance with the institutional guidelines of the Animal Care and
Use Committee of Renmin Hospital of Wuhan University (Wuhan, China)
and in accordance with the Guide for the Care of Laboratory Animals
published by the US National Institutes of Health (NIH publication
no. 85–23, revised in 1996). This study was approved by the ethics
committee of Wuhan University (Wuhan, China). A total of 60 male
C57BL/6 mice (age, 8–10 weeks; weight, 23.5–27.5 g) were purchased
from the Institute of Laboratory Animal Science, CAMS & PUMC
(Beijing, China). All animals were allowed to acclimatize to the
laboratory environment for 1 week, then randomly distributed to
either a sham surgery or AB group, which were respectively treated
without or with naringenin (normal diet containing ~100 mg/kg body
weight/day naringenin) for 7 weeks, beginning 1 week after AB
surgery. The dose of naringenin was administered according to the
protocol described in a previous study (10). Non-naringenin (vehicle) mice received
a normal diet of rodent chow. Subsequently, the C57BL/6 mice were
allocated at random into four groups: Sham + naringenin, AB +
naringenin, sham + vehicle and AB + vehicle (n=15 per group). AB
was performed as described previously (11). At 8 weeks after surgery, the animals
were euthanized by cervical dislocation while anesthetized with
1.5% isoflurane, and the hearts were dissected and weighed to
calculate the heart weight/body weight (HW/BW, in mg/g), heart
weight/tibia length (HW/TL, in mg/mm) and lung weight/body weight
(LW/BW, in mg/g) ratios in the naringenin-treated and
vehicle-treated mice. All surgeries and analyses were performed in
a blinded manner.
Echocardiography and hemodynamics
Transthoracic echocardiography and hemodynamic
analysis were performed according to the protocol described in a
previous study (12). The animals
were subjected to echocardiographic and pressure-volume analyses at
8 weeks after surgery. For echocardiography measurements, mice was
anesthetized with 1.5% isoflurane, and measurements were obtained
using a MyLab™ 30CV ultrasound system (Esaote SpA, Genoa, Italy)
equipped with a 10-MHz linear array ultrasound transducer. The left
ventricle (LV) dimensions were assessed in a parasternal short-axis
view during systole or diastole. The LV end-systolic diameter
(LVESD), LV end-diastolic diameter (LVEDD) and wall thickness were
obtained from the smallest or largest area of the LV. In the
process of hemodynamic measurements, a microtip catheter transducer
(Millar, Inc., Houston, TX, USA) was inserted into the right
carotid artery and advanced into the LV of mice anesthetized with
1.5% isoflurane. The signals were continuously recorded using a
Millar Pressure-Volume system (Millar, Inc.), and the maximal rate
of pressure development (dP/dtmax) and minimal rate of
pressure decay (dP/dtmin) were processed using PVAN data
analysis software (Millar, Inc.).
Histological analysis
The hearts of the mice were excised, arrested in
diastole with 10% KCl, weighed, placed in 10% formalin and embedded
in paraffin. The hearts were cut transversely close to the apex to
visualize the left and right ventricles. Sections of the heart (4–5
mm) were obtained and mounted onto slides, stained with hematoxylin
and eosin (H&E; Baso Diagnostics, Inc., Zhuhai, China) for
histopathological analysis. For collagen deposition determination,
tissue sections were stained with 0.1% picro-sirius red (PSR) red
satin solution (Dechuang, Inc., Beijing, China) for 1 h at room
temperature, then washed in 0.5% acetic acid for 2 min and
visualized by light microscopy (ECLIPSE 80i; Nikon Corporation,
Tokyo, Japan). For myocyte cross-sectional area examination,
sections were stained with H&E. A single myocyte per field was
observed using the Image-Pro Plus 6.0 quantitative digital image
analysis system (Media Cybernetics, Inc., Rockville, MD, USA). The
outline of 100–200 myocytes in the LV was analyzed for each
group.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) and western blot analysis
In order to investigate the relative mRNA expression
levels of atrial natriuretic peptide (ANP), B-type natriuretic
peptide (BNP), β-myosin heavy chain (β-MHC), transforming growth
factor-β (TGF-β), connective tissue growth factor (CTGF), collagen
Iα and collagen IIIα, total mRNA was extracted from frozen,
pulverized LV tissue using TRIzol reagent (cat. no. 15596026;
Invitrogen Life Technologies, Carlsbad, CA, USA). cDNA was
synthesized from 2 mg total RNA using oligo (dT) primers and an
RT-for-PCR kit (cat. no. 04896866001; Roche Diagnostics GmbH,
Mannheim, Germany). PCR amplifications were performed using a
LightCycler 480 SYBR Green Master Mix (cat. no. 04896866001; Roche
Diagnostics GmbH). The cycling conditions for PCR were as follows:
10 sec denaturation at 95°C, 20 sec annealing at 60°C and 20 sec at
72°C extension. The results were normalized against the expression
levels of glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
In order to determine the activation state of MAPK
and PI3K/Akt signaling, cardiac tissues were lysed in RIPA lysis
buffer. Subsequently, the protein concentration of all samples was
measured using a bicinchoninic assay kit (cat. no. 23227; Thermo
Fisher Scientific, Waltham, MA, USA). Tissue lysate samples (50 mg)
were subjected to 10% SDS-PAGE, and subsequently transferred to
polyvinylidene difluoride membranes (cat. no. IPFL00010; EMD
Millipore, Billerica, MA, USA). The membranes were blocked with 5%
non-fat milk and then incubated overnight at 4°C with the following
rabbit primary antibodies against: Phosph-PI3K p85Tyr458/p55Tyr199
(4288), total-PI3K p85 (4257), phospho-AktSer473 (4060), total-Akt
(4691), phospho-glycogen synthase kinase 3βSer9 (GSK3β) (9322),
total-GSK3β (9315), phospho-extracellular signal-regulated kinase
(ERK)1/2Thr202/Tyr204 (4370), total-ERK1/2 (4695),
phospho-p38Thr180/Tyr182 (4511), total-p38 (9212),
phospho-JNK1/2Thr183/Tyr18 (4668), total-JNK1/2 (9285) (1:1,000;
Cell Signaling Technology, Inc., Danvers, MA, USA). In addition,
rabbit anti-GAPDH antibody (1:1,000; MB001) was purchased from
Bioworld Technology, Inc. (St. Louis Park, MN, USA). The blots were
then scanned using a two-color infrared imaging system (Odyssey;
LI-COR Biosciences, Lincoln, NE, USA). Specific protein expression
levels were normalized against those of GAPDH protein for total
tissue lysates.
Statistical analysis
Data are expressed as the mean ± standard deviation.
Differences among groups were determined by two-way analysis of
variance followed by a post-hoc Tukey's test. Comparisons between
two groups were performed using an unpaired Students t-test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Naringenin protects against pressure
overload-induced cardiac hypertrophy
To determine the role of naringenin in cardiac
remodeling, hypertrophic responses were evaluated using
histological analysis and RT-qPCR. H&E staining of gross heart
tissue confirmed the protective effect of naringenin against
cardiac hypertrophy. H&E staining of gross heart tissue
indicated that the increase in the cardiomyocyte cross-sectional
area following AB was inhibited by naringenin (Fig. 1A). At 8 weeks of AB treatment, mice
in the AB + vehicle group exhibited significant increases in the
HW/BW, HW/TL and LW/BW ratios compared with the sham + vehicle
group. Administration of naringenin evidently decreased the HW/BW
and HW/TL ratios following AB (P<0.05), indicating the
attenuation of cardiac hypertrophy; however, no statistically
significant difference was detected in the LW/BW ratio (P>0.05)
(Fig. 1B). Furthermore, in mice
subjected to AB, the mRNA expression levels of hypertrophic
markers, including ANP, BNP and β-MHC, was mitigated by naringenin,
as determined by RT-qPCR (P<0.05) (Fig. 1C).
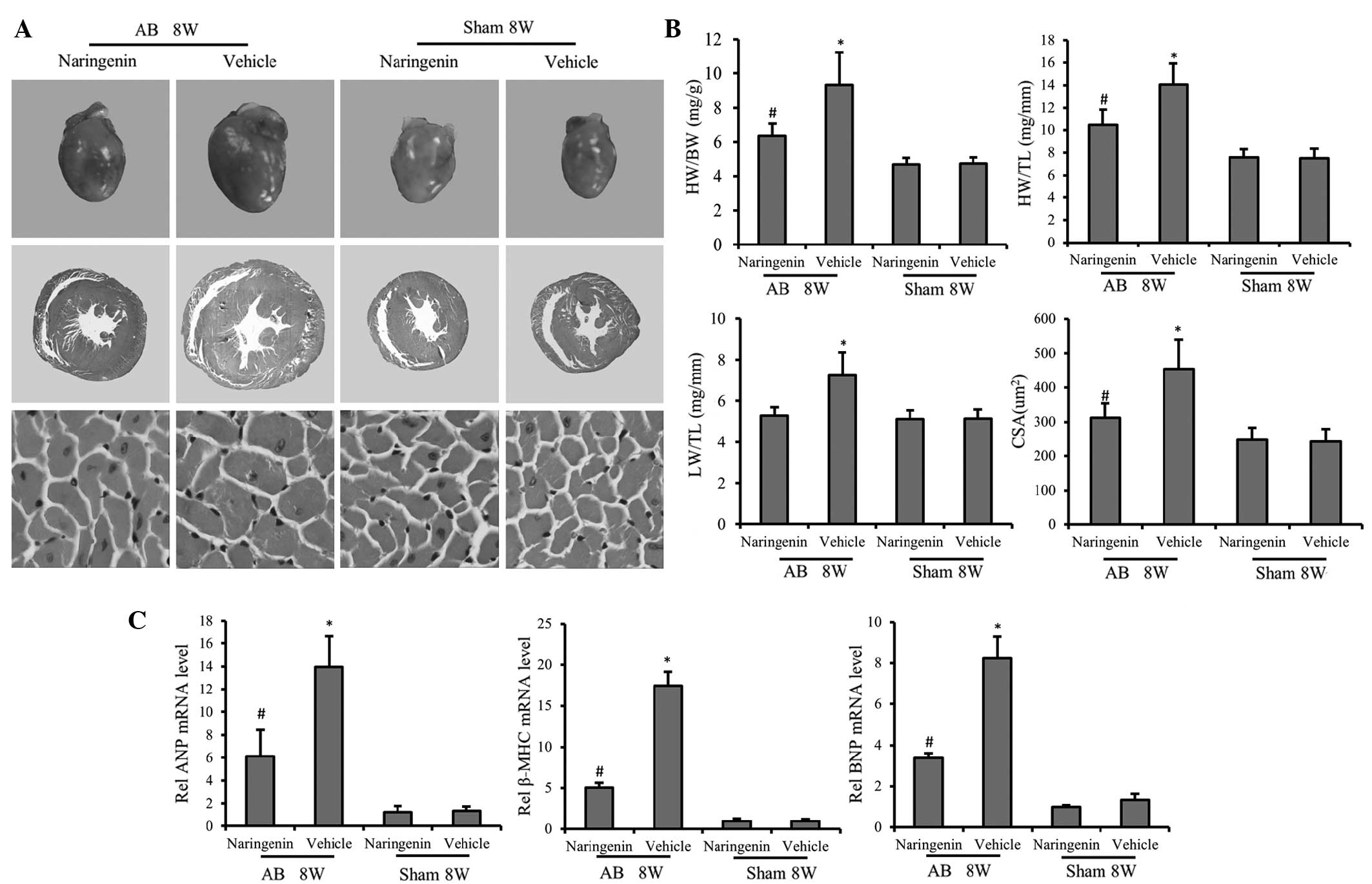 | Figure 1.Effect of naringenin on cardiac
hypertrophy. (A) Gross hearts (top), whole heart section (middle;
magnification, ×10) and cardiomyocyte cross-sectional area (bottom;
magnification, ×400) stained with hematoxylin and eosin at 8 weeks
after AB surgery. Naringenin treatment alleviated cardiac
hypertrophy and decreased the cardiomyocyte cross-sectional area
following AB surgery. (B) Results of the HW/BW, LW/BW and HW/TL
ratios, and myocyte CSA of the indicated groups. (C) Expression of
transcripts for ANP, BNP and β-MHC of the indicated groups were
determined by reverse transcription-polymerase chain reaction
analysis (fold changes). *P<0.05 vs. corresponding sham group.
#P<0.05 vs. AB + vehicle group. AB, aortic banding;
8W, 8 weeks; ANP, atrial natriuretic peptide; β-MHC, β-myosin heavy
chain; BNP, B-type natriuretic peptide; HW, heart weight; BW, body
weight; TL, tibial length; CSA, cross-sectional area. |
Naringenin improves impaired cardiac
function following AB
To evaluate the effect of naringenin in the
progression of cardiac dysfunction, AB or sham surgery was
performed. After 8 weeks, the chamber diameter, wall thickness and
function of the LV were assessed by echocardiography. No
statistically significant differences were detected between the
vehicle and naringenin-treated mice that underwent the sham
surgery. However, following AB surgery, the naringenin-treated mice
exhibited alleviated cardiac hypertrophy and cardiac dysfunction
compared with the vehicle mice, which was determined by measuring
the LVESD, LVEDD, fractional shortening and ejection fraction
(P<0.05) (Fig. 2A and B). The
dP/dtmax and dP/dtmin, analyzed by
pressure-volume loop, revealed that the naringenin treatment
exerted a beneficial effect on the hemodynamic function of LV
(Fig. 2C).
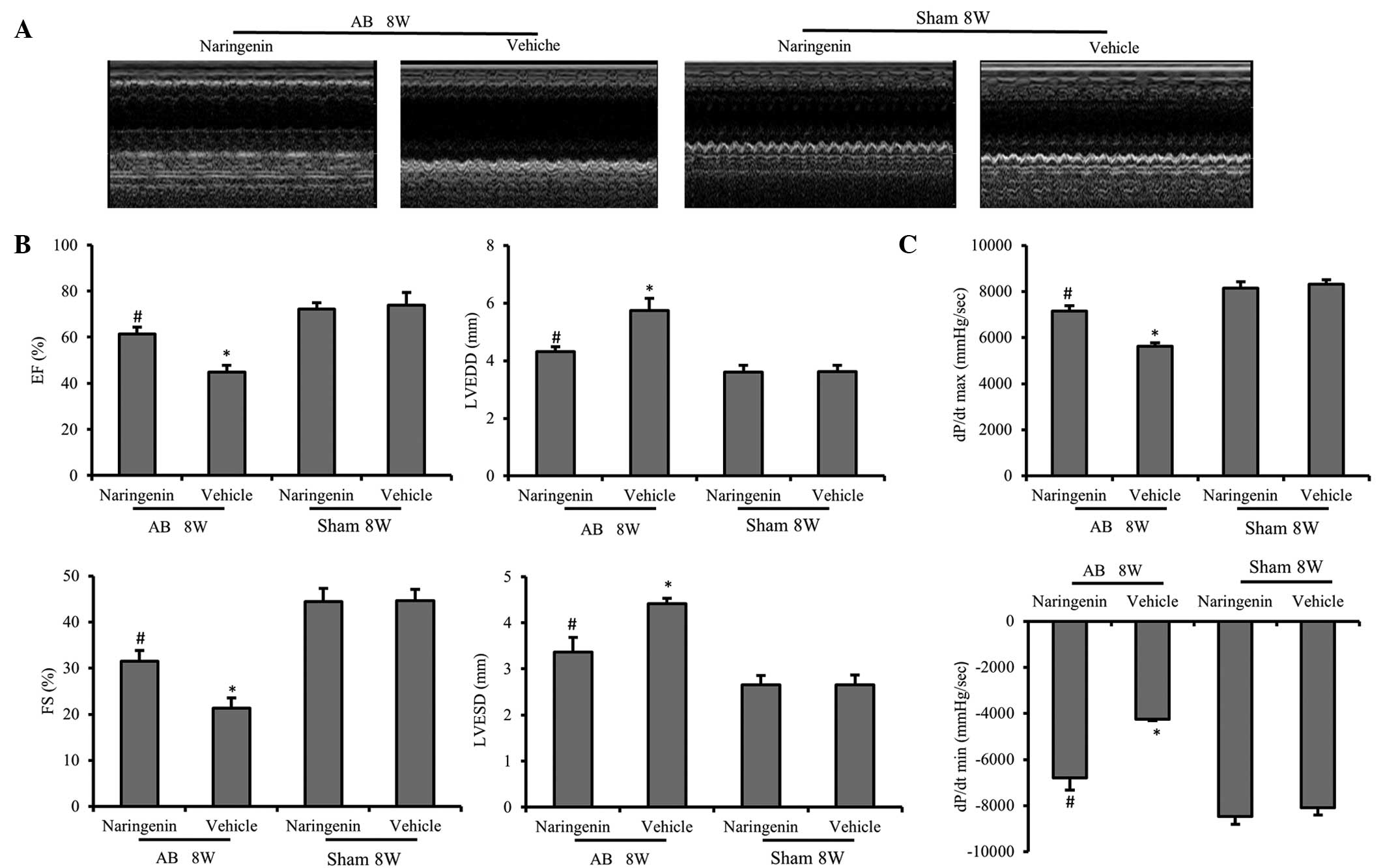 | Figure 2.Echocardiography and pressure-volume
analysis results indicating that naringenin improves the cardiac
function. (A) Representative M-mode images of AB and sham groups.
(B) Naringenin attenuated pressure overload-induced increased LV
diameters, including the LVESD, LVEDD, FS and EF. (C) Cardiac
function data of hemodynamic parameters including
dp/dtmax and dp/dtmin. *P<0.05 vs.
corresponding sham group. #P<0.05 vs. AB + vehicle
group. AB, aortic banding; EF, ejection fraction; LVEDD, left
ventricular (LV) end-diastolic diameter; LVESD, LV end-systolic
diameter; FS, fractional shortening; dp/dtmax, maximal
rate of pressure development; dp/dtmin, minimal rate of
pressure decay |
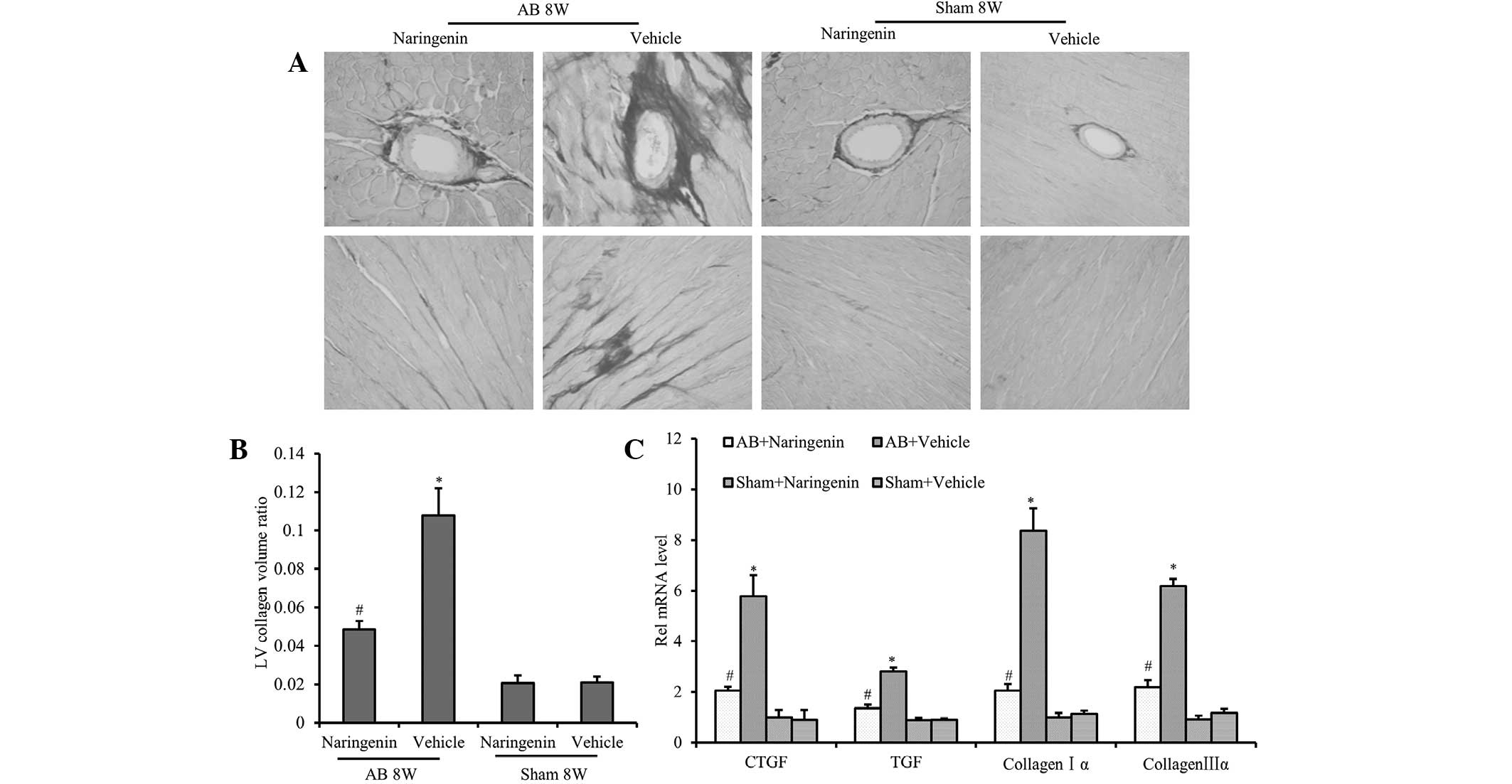
Naringenin alleviates the fibrotic
response induced by pressure overload
To clarify the effects of naringenin on perivascular
and interstitial fibrosis, cardiac interstitial collagen deposition
was detected using PSR staining. The results demonstrated that the
fibrotic area ratio was decreased significantly in mice that
received naringenin treatment following AB surgery (P<0.05).
However, there was no significant difference in the extent of
cardiac fibrosis in the sham-operated mice after 8 weeks
(P>0.05) (Fig. 3A and B). To
further elucidate the effect of naringenin on collagen synthesis,
the mRNA expression levels of TGF-β, CTGF, collagen Iα and collagen
IIIα, which are known mediators of fibrosis, were analyzed.
Compared with the vehicle group, decreased expression levels of
TGF-β, CTGF, collagen Iα and collagen IIIα were detected in the
naringenin group following the AB surgery (P<0.05) (Fig. 3C).
Naringenin attenuates cardiac
hypertrophy by inhibiting the activation of JNK, ERK and PI3K/Akt
signaling pathways
To elucidate the molecular mechanisms by which
naringenin mediates cardiac hypertrophy, the activation states of
the MAPK and PI3K/Akt signaling pathways were evaluated. The
results indicated that the upregulation of phospho-JNK and
phospho-ERK was mitigated in mice that received naringenin
treatment following the AB surgery (P<0.05). However, the level
of phospho-p38 was unchanged (P>0.05) (Fig. 4A and B). Furthermore, the role of the
PI3K/Akt signaling pathway, a key mechanism that regulates the
progression of pathological cardiac hypertrophy, was investigated.
Naringenin also appeared to decrease the expression of
phosphorylated Akt and its downstream target phospho-GSK3β
subsequent to pressure overload (P<0.05) (Fig. 4C and D).
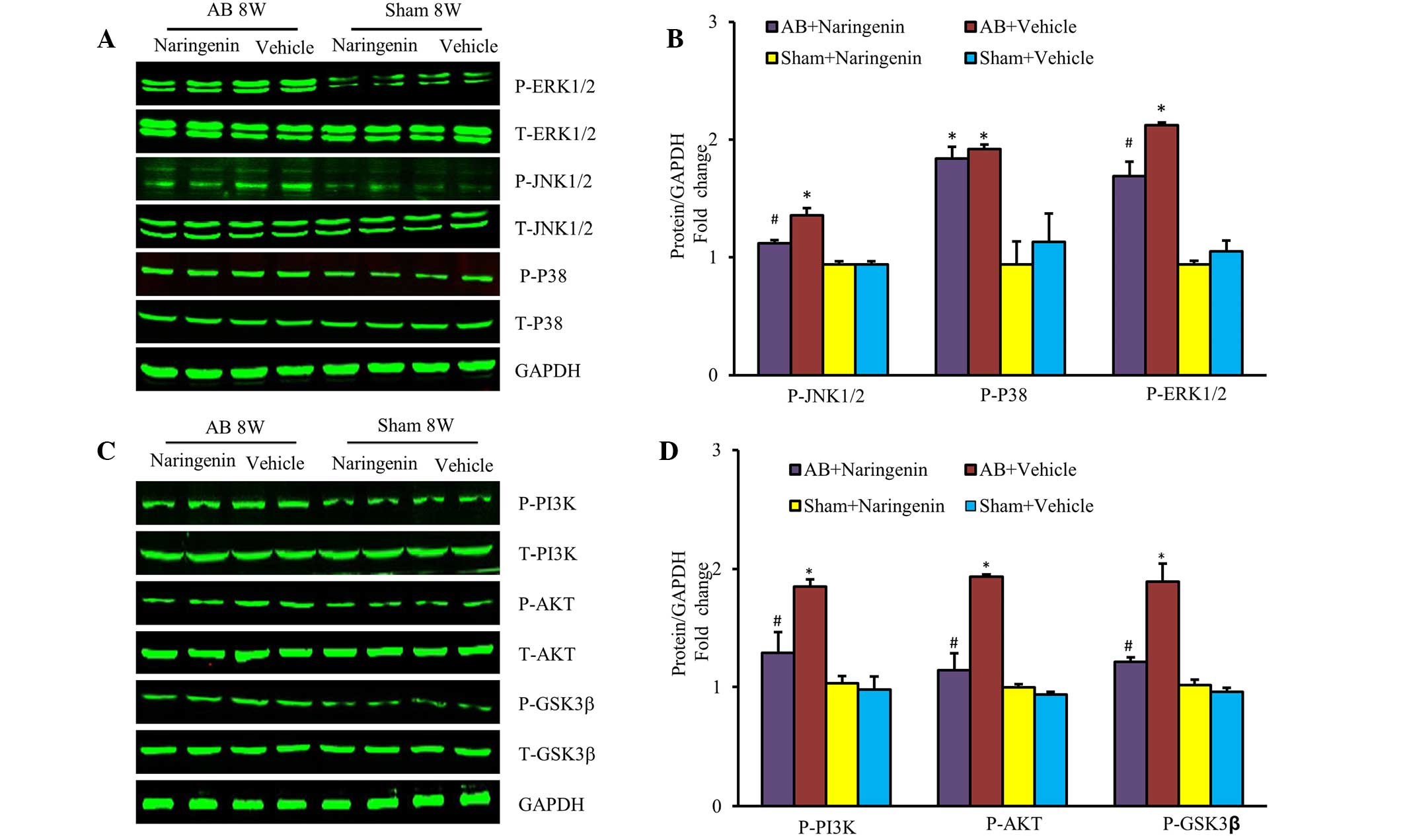 | Figure 4.Effects of naringenin on
mitogen-activated protein kinases and PI3K/AKT signalling in
response to hypertrophic stimuli. (A) Representative and (B)
quantitative expression of phosphorylated and total ERK1/2, JNK1/2,
p38, and the effects of naringenin on indicated groups. (C)
Representative and (D) quantitative blots of phosphorylated and
total PI3K, Akt, GSK3β in the heart tissues of mice in the
indicated groups, with GAPDH used as the loading control.
*P<0.05 vs. corresponding sham group. #P<0.05 vs.
AB + vehicle group. AB, aortic banding; ERK, extracellular
signal-regulated kinase; JNK, c-Jun N-terminal kinase; PI3K,
phosphoinositide 3-kinase; Akt, protein kinase B; GSK3β, glycogen
synthase kinase 3β; GAPDH, glyceraldehyde-3-phosphate
dehydrogenase. |
Discussion
Naringenin is widely used in traditional Chinese
medicine due to its multiple pharmacological effects, including
anti-inflammatory (5),
anti-hyperlipidemic, anti-thrombotic and anti-atherosclerotic
properties. Naringenin promotes endothelium-independent
vasorelaxing effects on rat aortic rings pre-contracted with
noradrenaline (9), and is able to
alleviate the myocardial ischemia/reperfusion injury (10). A previous study reported that
naringenin is able to protect against doxorubicin-induced cardiac
toxicity, due to its radical-scavenging and iron-chelating
properties (6). To the best of our
knowledge, the present study demonstrated for the first time that
naringenin is able to attenuate pressure overload-induced cardiac
hypertrophy. Furthermore, the results indicated that naringenin may
improve cardiac function and attenuate the interstitial
fibrosis.
Cardiac hypertrophy is characterized by the abnormal
enlargement of cardiomyocytes, originating from an increase in
myocyte size and the proliferation of non-muscle cells (13). Numerous clinical studies have
indicated that sustained cardiac hypertrophy is a maladaptive
process, ultimately leading to decompensated heart failure
(14). Characterization of the
pathogenesis of cardiac hypertrophy may provide novel approaches
for the prevention of heart failure progression. Blockage of the
MAPK signaling pathway, downstream of which a variety of
intracellular transcription factors are phosphorylated during the
reprogramming of cardiac gene expression, prevents the progression
of cardiac hypertrophy (15). In
addition, the ERK pathway serves a critical function in cardiac
hypertrophy, and may be activated by mitogen-activated protein
kinase kinase (known as MEK or MAPKK)-dependent phosphorylation
(16). JNKs are specific transducers
of the stress response, and are known as stress-activated protein
kinases (17). In the heart, JNK
activity is upregulated following pressure overload (18). However, Lei et al (19) reported that the expression of p38
MAPK was upregulated transiently in transverse AB-induced pressure
overload in mice. However, the molecular mechanism by which
naringenin mitigates cardiac hypertrophy remain unclear. Therefore,
in the present study the activation of potential signaling
pathways, including the MAPK and PI3K/Akt pathways, was evaluated.
The results of the present study demonstrate that naringenin is
able to mitigate the activation of the ERK and JNK signaling
pathways. PI3K/Akt is another crucial signaling pathway in the
process of cardiac hypertrophy. Various studies have indicated that
the engagement of the p85SH2 domains of PI3K by pTyr relieved the
p85-mediated inhibition of p110 isoforms, led to the activation of
class I PI3Ks (p110α, p110δ, p110β) and always resulted in Akt
activation (20). The short-term
activation of Akt known to be caused by physiological hypertrophy
and through such pathways may be cardioprotective, despite causing
mild heart enlargement; however, the long-term activation promoted
pathological cardiac hypertrophy and heart failure (21). Inhibition of GSK3β, downstream of
Akt, is required for compensation of pressure overload (22), and has been shown to be a negative
regulator of cardiac hypertrophy (23). Notably, naringenin attenuates the
activation of Akt in the progression of cardiac hypertrophy.
Fibrosis is another integral feature of cardiac
hypertrophy, which is characterized by the disproportionate
expression of the extracellular matrix and accumulation of
fibrillar collagen. The primary collagen types in the heart are
type I and III, which together account for >90% of the total
collagen and are the predominant contributors to interstitial
fibrosis in the progression of heart failure (24). The present results indicate that the
mRNA expression levels of TGF-β1, CTGF, collagen Iα and collagen
IIIα were increased following AB, and that naringenin evidently
alleviated this upregulation. In addition, the results of the PSR
staining suggested that naringenin attenuates the interstitial and
perivascular fibrosis induced by pressure overload (25). Notably, naringenin has the
pharmacological effect of normalizing the expression of
pro-fibrotic genes, indicating that naringenin may be effective in
attenuating extracellular remodeling following pressure overload.
These effects require verification by additional studies.
In conclusion, the results of the present study
demonstrated that naringenin exerts a pharmacological effect
against the progression of cardiac hypertrophy induced by pressure
overload. Inhibition of the JNK, ERK and PI3K/Akt signaling
pathways by naringenin may serve a crucial function in the
mitigation of the development of cardiac hypertrophy in structure
remodeling and function alteration induced by pressure overload.
This anti-fibrotic function is mediated by the downregulation of
interstitial fibrosis gene expression, including that of TGF-β1,
CTGF, collagen Iα and collagen IIIα, which occur in response to
pressure overload. These results partially elucidate the
pharmacological effects of naringenin and the pathways involved in
its protective role, which may provide a novel pharmacotherapeutic
strategy for the treatment of cardiac hypertrophy in pressure
overload and limit the progression of heart failure.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (no. 81270303 and 81300104),
the Specialized Research Fund for the Doctoral Program of Higher
Education of China (no. 20130141120042) and the Natural Science
Foundation of Hubei Province, China (no. 2013CFB303).
References
|
1
|
Balakumar P and Jagadeesh G: Multifarious
molecular signaling cascades of cardiac hypertrophy: Can the muddy
waters be cleared? Pharmacol Res. 62:365–383. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gjesdal O, Bluemke DA and Lima JA: Cardiac
remodeling at the population level-risk factors, screening and
outcomes. Nat Rev Cardiol. 8:673–685. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hellawell JL and Margulies KB: Myocardial
reverse remodeling. Cardiovasc Ther. 30:172–181. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
van Berlo JH, Maillet M and Molkentin JD:
Signaling effectors underlying pathologic growth and remodeling of
the heart. J Clin Invest. 123:37–45. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yuan Y, Zong J, Zhou H, Bian ZY, Deng W,
Dai J, Gan HW, Yang Z, Li H and Tang QZ: Puerarin attenuates
pressure overload-induced cardiac hypertrophy. J Cardiol. 63:73–81.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Middleton E Jr and Kandaswami C: Effects
of flavonoids on immune and inflammatory cell functions. Biochem
Pharmacol. 43:1167–1169. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Oshipura KJ, Hu FB, Manson JE, Stampfer
MJ, Rimm EB, Speizer FE, Colditz G, Ascherio A, Rosner B,
Spiegelman D and Willett WC: The effect of fruit and vegetable
intake on risk for coronary heart disease. Ann Intern Med.
134:1106–1114. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dauchet L, Ferrières J, Arveiler D,
Yarnell JW, Gey F, Ducimetière P, Ruidavets JB, Haas B, Evans A,
Bingham A, et al: Frequency of fruit and vegetable consumption and
coronary heart disease in France and Northern Ireland: The PRIME
study. Br J Nutr. 92:963–972. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Saponara S, Testai L, Iozzi D, Martinotti
E, Martelli A, Chericoni S, Sgaragli G, Fusi F and Calderone V:
(+/−)-Naringenin as large conductance Ca(2+)-activated K+ (BKCa)
channel opener in vascular smooth muscle cells. Br J Pharmacol.
149:1013–1021. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Testai L, Martelli A, Marino A,
D'Antongiovanni V, Ciregia F, Giusti L, Lucacchini A, Chericoni S,
Breschi MC and Calderone V: The activation of mitochondrial BK
potassium channels contributes to the protective effects of
naringenin against myocardial ischemia/reperfusion injury. Biochem
Pharmacol. 85:1634–1643. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yan L, Huang H, Tang QZ, Zhu LH, Wang L,
Liu C, Bian ZY and Li H: Breviscapine protects against cardiac
hypertrophy through blocking PKC-alpha-dependent signaling. J Cell
Biochem. 109:1158–1171. 2010.PubMed/NCBI
|
|
12
|
Zhou H, Bian ZY, Zong J, Deng W, Yan L,
Shen DF, Guo H, Dai J, Yuan Y, Zhang R, et al: Stem cell antigen 1
protects against cardiac hypertrophy and fibrosis after pressure
overload. Hypertension. 60:802–809. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Carreño JE, Apablaza F, Ocaranza MP and
Jalil JE: Cardiac hypertrophy: Molecular and cellular events. Rev
Esp Cardiol. 59:473–486. 2006.(In Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Frey N and Olson EN: Cardiac hypertrophy:
The good, the bad and the ugly. Annu Rev Physiol. 65:45–79. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lorenz K, Schmitt JP, Vidal M and Lohse
MJ: Cardiac hypertrophy: Targeting Raf/MEK/ERK1/2-signaling. Int J
Biochem Cell Biol. 41:2351–2355. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lorenz K, Schmitt JP, Schmitteckert EM and
Lohse MJ: A new type of ERK1/2 autophosphorylation causes cardiac
hypertrophy. Nat Med. 15:75–85. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Paul A, Wilson S, Belham CM, Robinson CJ,
Scott PH, Gould GW and Plevin R: Stress-activated protein kinases:
Activation, regulation and function. Cell Signal. 9:403–410. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sopontammarak S, Aliharoob A, Ocampo C,
Arcilla RA, Gupta MP and Gupta M: Mitogen-activated protein kinases
(p38 and c-Jun NH2-terminal kinase) are differentially regulated
during cardiac volume and pressure overload hypertrophy. Cell
Biochem Biophys. 43:61–76. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lei B, Chess DJ, Keung W, O'Shea KM,
Lopaschuk GD and Stanley WC: Transient activation of p38 MAP kinase
and up-regulation of Pim-1 kinase in cardiac hypertrophy despite no
activation of AMPK. J Mol Cell Cardiol. 45:404–410. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vanhaesebroeck B, Guillermet-Guibert J,
Graupera M and Bilanges B: The emerging mechanisms of
isoform-specific PI3K signalling. Nat Rev Mol Cell Biol.
11:329–341. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chaanine AH and Hajjar RJ: AKT signalling
in the failing heart. Eur J Heart Fail. 13:825–829. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Badorff C, Ruetten H, Mueller S, Stahmer
M, Gehring D, Jung F, Ihling C, Zeiher AM and Dimmeler S: Fas
receptor signaling inhibits glycogen synthase kinase 3 beta and
induces cardiac hypertrophy following pressure overload. J Clin
Invest. 109:373–381. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Haq S, Choukroun G, Kang ZB, Ranu H,
Matsui T, Rosenzweig A, Molkentin JD, Alessandrini A, Woodgett J,
Hajjar R, et al: Glycogen synthase kinase-3beta is a negative
regulator of cardiomyocyte hypertrophy. J Cell Biol. 151:117–130.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Creemers EE and Pinto YM: Molecular
mechanisms that control interstitial fibrosis in the
pressure-overloaded heart. Cardiovasc Res. 89:265–272. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hutchinson KR, Guggilam A, Cismowski MJ,
Galantowicz ML, West TA, Stewart JA Jr, Zhang X, Lord KC and
Lucchesi PA: Temporal pattern of left ventricular structural and
functional remodeling following reversal of volume overload heart
failure. J Appl Physio. 111:1778–1788. 2011. View Article : Google Scholar
|