Introduction
Inflammation is of importance in the highly complex
immune response mounted to defend against harmful stimuli,
including pathogens, damaged cells or irritants. Macrophages, as
critical participants in the inflammatory process, directly
counteract the aforementioned stimuli (1,2). Under
inflammatory conditions, enzymes, cytokines and chemokines, in
addition to signaling proteins at the site of the infected tissues
and cells, are secreted by macrophages, causing inflammatory cells
to migrate to sites of inflammation in order to resolve the
abnormal conditions (1,2). The model most commonly used to
investigate induced inflammation is the stimulation of macrophages
by lipopolysaccharide (LPS) obtained from gram-negative bacteria
(1). The binding of LPS to its
cognate receptors, including Toll-like receptors on the mammalian
cell surface, activates several signaling cascades that drive the
expression of pro-inflammatory mediators and cytokines, including
nitric oxide (NO), prostaglandin E2, tumor necrosis
factor-α (TNF-α) and interleukin (IL)-1β (2,3).
Additionally, excessive inflammatory responses may
result in decreased expression of anti-inflammatory cytokines
(1,2,4,5). The pathogenesis of autoimmune diseases,
which are characterized by chronic inflammation, may be influenced
by pro- and anti-inflammatory factors, as well as dysregulation of
inflammatory immune responses (5,6).
Therefore, the utilization of different therapeutic strategies may
be therapeutically beneficial for the treatment of inflammatory
diseases; for example, bioactive agents could be used to inhibit
the production of inflammatory factors by macrophages (4–6).
Subsequent to stimulation with LPS, nuclear
factor-κB (NF-κB) is activated as a result of the activation of the
inhibitor of κB (IκB)-kinase complex inhibitor (7,8).
Inhibitor of IκB phosphorylates IκB, causing IκB proteasomal
degradation and the release of NF-κB (7,8). The
liberated dimeric NF-κB then translocates to the nucleus and
activates the transcription of pro-inflammatory target genes that
encode regulatory proteins. This leads to physiological responses,
including inflammatory or immune responses (7,8).
Downstream targets of the LPS-induced inflammatory cascades in
macrophages are members of the mitogen-activated protein kinase
(MAPK) family, including extracellular signal-regulated kinase
(ERK), c-Jun NH2-terminal kinase (JNK) and p38 MAPK
(2,3,7). When
macrophages are stimulated with LPS, MAPKs are activated to produce
inflammatory factors through the activation of multiple downstream
signaling events (9,10). Therefore, targeting the NF-κB and
MAPK signaling pathways is considered an attractive therapeutic
strategy for the development of anti-inflammatory therapeutic
agents.
Ginseng, the root of Panax ginseng C.A. Meyer
(Araliaceae), is a herbal medicine that has been shown to exhibit a
variety of therapeutic effects, including neuroprotective,
immunomodulating, anti-cancer and antioxidant activities (9). As part of traditional folk medicine,
ginseng has been a popular plant-based medicine for 2,000 years in
East Asian countries, and remains a popular natural medicine
worldwide (11–13). The major active constituents of
ginseng are the triterpenoid saponins, also known as total saponins
extracted from ginseng (TSG), which have a four-ring, steroid-like
structure with attached sugar moieties (14–16). TSG
are the primary molecules responsible for the effects of ginseng.
They possess pharmacological properties, including
anti-inflammatory, antioxidant and anti-cancer effects (11–13). In
addition, Kim et al (17)
suggested that TSG may suppress NO production in
LPS/interferon-γ-activated RAW 246.7 macrophages by inhibiting
inducible NO synthase (iNOS) expression. Furthermore, TSG has been
found to inhibit LPS-induced expression of iNOS, matrix
metalloproteinase-9 and pro-inflammatory cytokines in BV2
microglial cells, which is associated with the inactivation of
NF-κB and MAPK signaling pathways (18). A recent study also demonstrated that
TSG-associated recovery from LPS-induced depression-like behavior
was associated with decreased production of various
pro-inflammatory cytokines in LPS-challenged mice and RAW 264.7
cells (19). However, the
anti-inflammatory mechanisms in macrophages have yet to be
elucidated.
Therefore, in the present study, the
anti-inflammatory effects of TSG were evaluated using an
LPS-stimulated RAW 264.7 macrophage cell model. It was identified
that TSG exerted curative effects on anti-inflammatory activity by
reducing iNOS production, and TNF-α and IL-1β expression. The
effects of TSG on the NF-κB and MAPK signaling pathways were also
investigated in the current study in order to elucidate the
associated inhibitory mechanism. The results provide evidence in
favor of the use of TSG as a potential anti-inflammatory
supplement.
Materials and methods
Preparation of TSG
For the preparation of TSG, air-dried ginseng roots
were purchased from Dongeui University Hospital of Oriental
Medicine (Busan, Korea). TSG was separated and purified as
described by Sugimoto et al (20). Briefly, samples were extracted twice
with methanol (Sigma-Aldrich, St. Louis, MO, USA) by refluxing at
80°C for 2 h. The methanol extract was then suspended in water and
partitioned sequentially with n-hexane, chloroform, ethyl acetate
and n-butanol. Subsequently, the water-saturated n-butanol fraction
was evaporated to dryness in a vacuum. The recovered crude saponins
were loaded onto Diaion® HP-20/MCI GEL® CHP20P (Sigma-Aldrich), and
the sugar residues were removed with 40% methanol. The fractions
were eluted with 60–80% methanol, collected, and dried to obtain
TSG. TSG was then diluted in Dulbecco's modified Eagle medium
(DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham) MA, USA) to
0, 50, 100, 200 and 400 µg/ml, prior to use.
Cell culture
Mouse RAW 264.7 macrophages were purchased from the
American Tissue Culture Collection (Manassas, VA, USA). RAW 264.7
cells (5×105 cells/ml) were grown in DMEM supplemented
with 10% fetal bovine serum and 1% (v/v) penicillin (100
U/ml)/streptomycin (100 µg/ml) under humidified conditions (5%
CO2 at 37°C). In all experiments, the cells were treated
with various concentrations of TSG (0, 50, 100, 200 or 400 µg/ml)
for 1 h prior to exposure with 100 ng/ml LPS (Lipopolysaccharides
from Escherichia coli 026:B6; Sigma-Aldrich) for 24 h under
humidified conditions (5% CO2 at 37°C).
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT)
assay
Cell viability was measured based on the formation
of blue formazan, which is metabolized from colorless MTT
(Sigma-Aldrich) by mitochondrial dehydrogenases, enzymes that are
only active in live cells (18). RAW
264.7 cells (5×105 cells/ml) were seeded in a 96-well
plate. The cells were pretreated with various concentrations of TSG
for 1 h and then stimulated by 100 ng/ml LPS for 24 h. Following
incubation with TSG and LPS, the cultured media was replaced with
fresh media and the cells were incubated with 0.5 mg/ml MTT
solution for 3 h. The supernatant was then discarded and the
formazan blue, which was formed in the cells, was dissolved with
dimethyl sulfoxide (Sigma-Aldrich). The optical density was
measured at 540 nm with an ELISA plate reader (MRX; Dynatech
Laboratories, Chantilly, VA, USA). Cell viability was calculated as
the percentage of surviving cells over control cells (no TSG
added).
Measurement of NO production
Concentrations of NO in the culture supernatants
were determined by measuring the levels of nitrite, which is a
major stable product of NO, using Griess reagent (Sigma-Aldrich).
RAW 264.7 cells (5×105 cells/ml) were seeded in each
well of a 96-well plate. The cells were pretreated with the
indicated concentrations of TSG for 1 h and stimulated with 100
ng/ml LPS. Following an incubation period of 24 h, the supernatant
from each well was collected by centrifugation at 3,000 × g using
the Smart R17 Refrigerated Micro Centrifuge (Hanil BioMed, Inc.,
Gwangju, Korea) for 20 min at 4°C. The supernatant was mixed with
the same volume of Griess reagent for 10 min at room temperature in
the dark. Nitrite levels were determined using an ELISA plate
reader (MRX; Dynatech Laboratories) at 540 nm, and nitrite
concentrations were calculated by referencing a standard curve
generated by known concentrations of sodium nitrite (17).
Measurement of TNF-α and IL-1β
production
To measure the inhibitory efficacy of TSG on the
production of TNF-α and IL-1β, TNF-α and IL-1β ELISA kits (cat nos.
MTA00B and MLB00C, respectively) were purchased from R&D
Systems, Inc. (Minneapolis, MN, USA). The cell culture conditions
were the same as for the nitrite measurement assay. After
incubation with TSG and LPS for 24 h, the TNF-α and IL-1β
concentrations in cultured media were determined by a selective
ELISA kit, according to the manufacturer's protocol.
Reverse transcription-polymerase chain
reaction (RT-PCR)
The cells were incubated with TSG (400 µg/ml) alone
for 24 h, or pretreated with the various concentrations of TSG for
1 h prior to LPS stimulation (100 ng/ml) for 24 h. Total RNA was
isolated from cultured cells using TRIzol® reagent (Invitrogen;
Thermo Fisher Scientific). Contaminating DNA was removed by
treating the RNA samples with recombinant DNase I (DNA-free™ kit;
Thermo Fisher Scientific, Inc.) The isolated RNA (1 µg) was used
for cDNA synthesis using AccuPower® RT PreMix (Bioneer Corporation,
Daejeon, Korea) containing Moloney murine leukemia virus reverse
transcriptase. iNOS, IL-1β and TNF-α genes were amplified from the
cDNA by PCR (5331 MasterCycler Gradient; Eppendorf, Hamburg,
Germany). The PCR primers were as follows: iNOS forward,
5′-ATGTCCGAAGCAAACATCAC-3′ and reverse, 5′-TAATGTCCAGGAAGTAGGTG-3′;
IL-1β forward, 5′-GGGCTGCTTCCAAACCTTTG-3′ and reverse,
5′-GCTTGGGATCCACACTCTCC-3′; TNF-α forward,
5′-TCTCATCAGTTCTATGGCCC-3′ and reverse, 5′-GGGAGTAGACAAGGTACAAC-3′;
and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) forward,
5′-AGGCCGGTGCTGAGTATGTC-3′ and reverse, 5′-TGCCTGCTTCACCACCTTCT-3′
(Bioneer Corporation). The PCR reaction was initiated at 94°C for 2
min, followed by 31 cycles of 94°C for 30 sec, 30 sec annealing
temperature, 72°C for 30 sec, and a final extension step at 72°C
for 5 min. The annealing temperatures were 63°C for iNOS, IL-1β and
TNF-α, and 61°C for GAPDH. Following amplification, the PCR
products were separated by 1.5% agarose gel electrophoresis,
stained with ethidium bromide (Sigma-Aldrich) and visualized by
ultraviolet illumination.
Protein extraction and western blot
analysis
For total protein extraction, the cells were lysed
by incubating with a lysis buffer [25 mM Tris-Cl (pH 7.5), 250 mM
NaCl, 5 mM ethylene diaminetetra acetic acid, 1% NP-40, 1 mM
pheny-methylsulfonyl fluoride and 5 mM dithiothreitol;
Sigma-Aldrich] for 1 h at 4°C. Insoluble materials were discarded
by centrifugation at 13,000 × g using the Smart R17 Refrigerated
Micro Centrifuge (Hanil BioMed, Inc.) for 20 min at 4°C. In a
parallel experiment, nuclear and cytosolic proteins were separated
using nuclear extraction reagents (product no. 78833; Pierce
Biotechnology, Inc., Rockford, IL, USA), according to the
manufacturer's protocol. The protein concentration in the cell
lysate was determined using a DC™ Protein Assay (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Equal quantities of protein
were then separated by 10% sodium dodecyl sulfate-polyacrylamide
gel electrophoresis at 90 V for 2 h. Separated protein was
transferred to a nitrocellulose membrane (Schleicher and Schuell,
Keene, NH, USA) and subsequently blocked with Tris-buffered saline
(10 mM of Tris-Cl, pH 7.4) containing 0.5% Tween-20 (TBST) and 5%
nonfat dried milk for 1 h at room temperature. Subsequently, the
membranes were probed with rabbit anti-iNOS (1:1,000; sc-509),
−IL-1β (1:1,000; sc-7884), −p65 (1:500; sc-109), −IκB-α (1:500;
sc-847), −ERK (1:1,000; sc-154) and −p38 (1:1,000; sc-535), and
goat anti-lamin B (1:1,000; sc-6216) and −actin (1:1,000; sc-1615)
polyclonal antibodies, from Santa Cruz Biotechnology, Inc. (Dallas,
TX, USA), and rabbit anti-TNF-α (1:1,000; #3707), −JNK (1:1,000;
#9252S) and phosphorylated (p)-38 (1:500; #9211S) polyclonal
antibodies, and mouse anti-p-JNK (1:500; #9255) and −p-ERK (1:500;
#9106S) monoclonal antibodies, from Cell Signaling Technology, Inc.
(Danvers, MA, USA), overnight at 4°C. After 2 h blocking with 5%
non-fat milk in TBST (1.5 M NaCl, 20 mM Tris-Cl, 0.05% Tween-20, pH
7.4), the membranes were incubated with horseradish
peroxidase-conjugated anti-rabbit IgG secondary antibodies
(1:1,000; RPN4301; GE Healthcare Life Sciences, Chalfont, UK) at
room temperature for 2 h. Using an enhanced chemiluminescence
detection system (GE Healthcare Life Sciences), immunoreactive
bands were detected and exposed to X-ray film.
Statistical analysis
Data are presented as the mean ± standard deviation.
Differences in mean values between groups were analyzed by a
one-way analysis of variance followed by Dunnett's-test. P<0.05
was considered to indicate a statistically significant
difference.
Results
TSG inhibits LPS-induced NO production
in RAW 264.7 macrophages
To evaluate the anti-inflammatory activity of TSG,
the effect of TSG on NO production stimulated by LPS was initially
investigated in RAW 264.7 cells. Following pretreatment with
various concentrations of TSG for 1 h, RAW 264.7 cells were
stimulated with LPS for 24 h. Cell culture media were collected and
NO levels were quantified using the Griess test. The addition of
LPS to RAW 264.7 cells resulted in an increase in NO production
levels (Fig. 1). However, TSG was
observed to significantly suppress LPS-induced NO production in a
concentration-dependent manner (Fig.
1; P<0.05).
TSG downregulates LPS-induced iNOS
expression in RAW 264.7 macrophages
Following the observation that NO production was
inhibited by TSG, RT-PCR and western blot analysis were performed
to determine whether the inhibition of NO production by TSG in the
LPS-stimulated RAW 264.7 cells was associated with decreased levels
of iNOS mRNA and protein, as iNOS produces NO as a key mediator of
inflammation. In response to LPS treatment, mRNA expression of iNOS
was induced; however, pretreatment with TSG inhibited this
upregulation at a dose of 400 µg/ml (Fig. 2A). Similarly, pretreatment with TSG
markedly suppressed the protein expression levels of iNOS in a
concentration-dependent manner (Fig.
2B). The results indicate that TSG can downregulate NO
production in LPS-stimulated RAW 264.7 cells via inhibition of iNOS
expression at the transcriptional level.
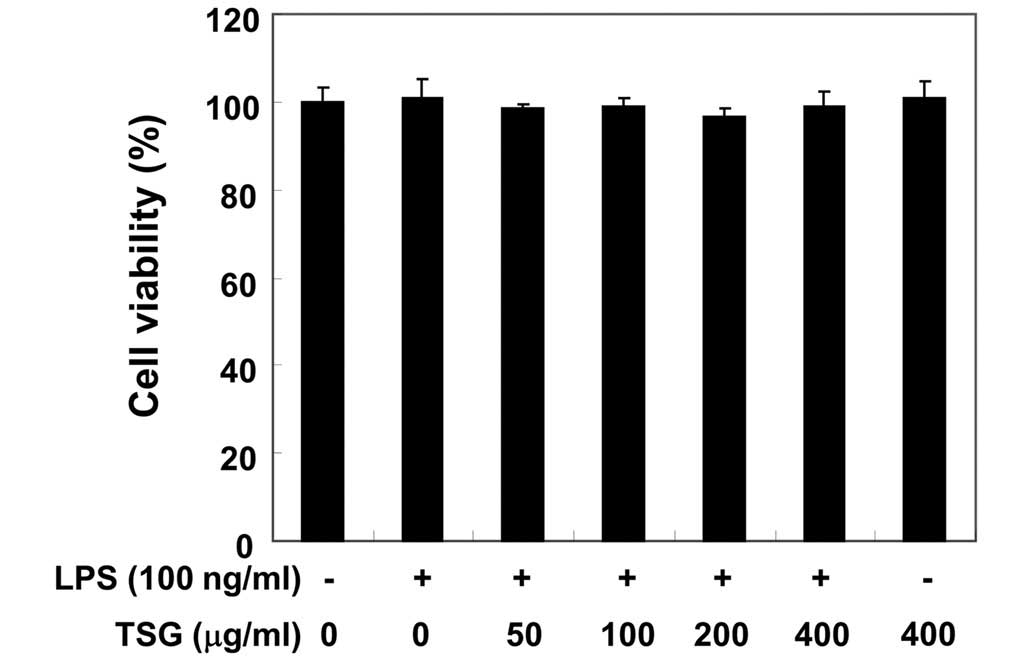
Effects of TSG on the viability of RAW
264.7 macrophages
In order to exclude the possibility that the
inhibition of NO production was due to cytotoxicity caused by TSG
treatment, MTT assays were performed in RAW 264.7 cells treated
with TSG for 24 h in the presence or absence of LPS (100 ng/ml). As
demonstrated in Fig. 3, at the same
concentrations of TSG (50–400 µg/ml) used to inhibit NO and its
production, TSG alone did not affect cell viability. Furthermore,
co-treatment with TSG and LPS also did not demonstrate any
cytotoxic effects. Thus, the results indicate that the inhibition
of NO production in LPS-stimulated RAW 264.7 cells was not a result
of the cytotoxic effects of TSG.
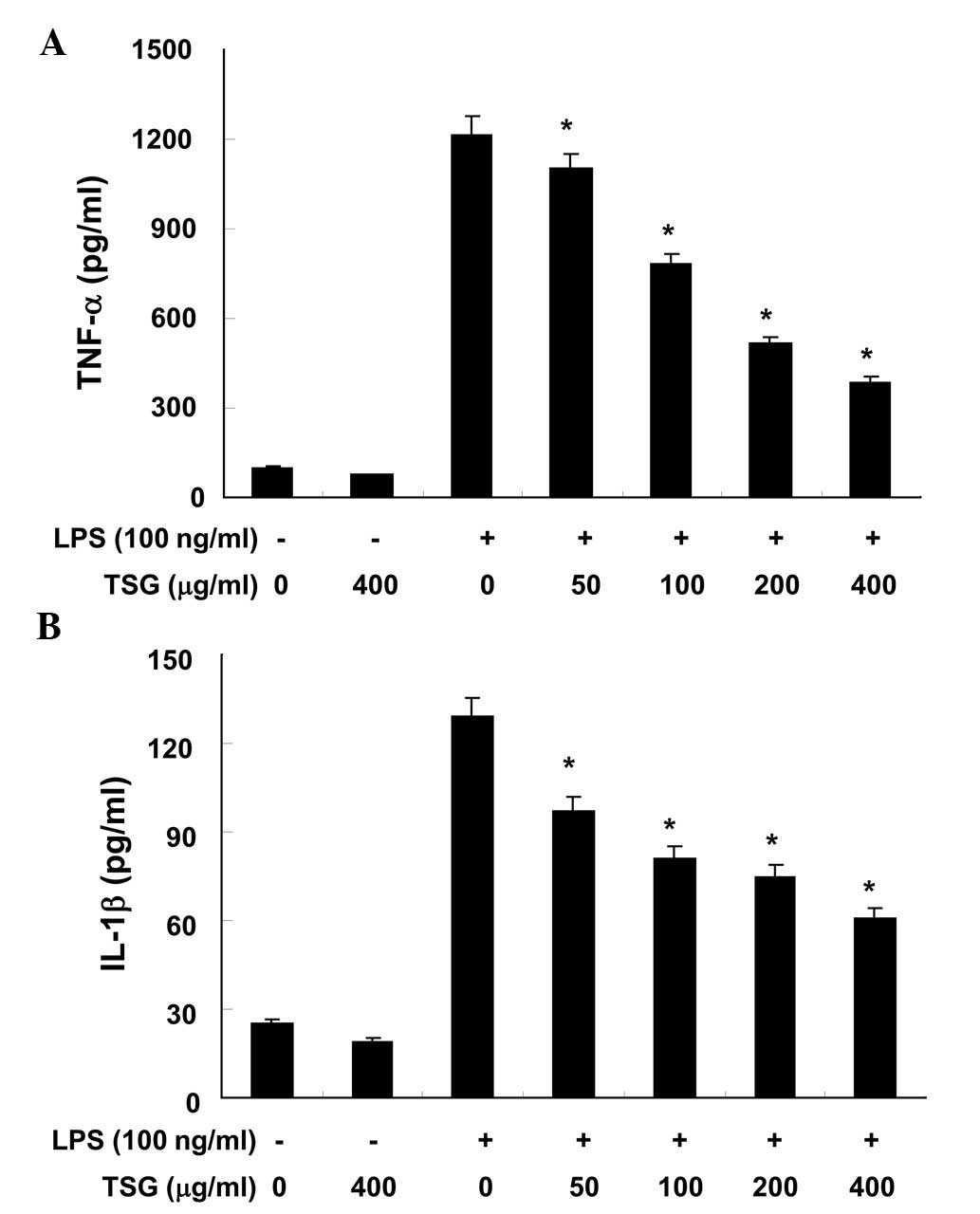
TSG reduces the production of
pro-inflammatory cytokines in LPS-stimulated RAW 264.7
macrophages
The effects of TSG on pro-inflammatory cytokines,
such as TNF-α and IL-1β, were analyzed using ELISA. As demonstrated
in Fig. 4A and B, when RAW 264.7
cells were treated with TSG alone, there were no significant
changes in the production levels of TNF-α or IL-1β, respectively
(P>0.05). However, production of the two cytokines were
increased in the culture media of LPS-stimulated RAW 264.7
macrophages, and these increases were significantly reduced by
treatment with TSG in a concentration-dependent manner (P<0.05;
Fig. 4).
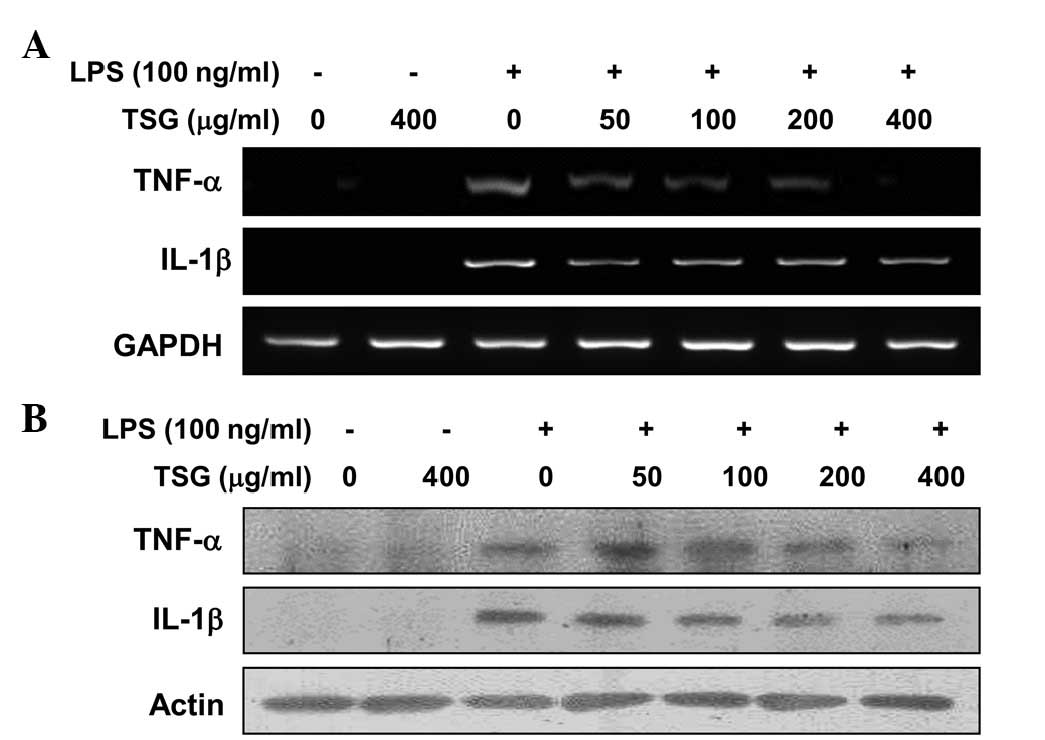
TSG downregulates LPS-induced TNF-α
and IL-1β mRNA and protein expression in RAW 264.7 macrophages
In a parallel experiment, RT-PCR and western blot
analyses were performed to determine whether TSG was able to
inhibit the expression of TNF-α and IL-1β cytokines at the
transcriptional or translational levels. As indicated in Fig. 5, treating RAW 264.7 macrophages with
different concentrations of TSG prior to LPS treatment resulted in
a dose-dependent decrease in the mRNA (Fig. 5A) and protein (Fig. 5B) levels of the two cytokines. Thus,
the results indicate that TSG also inhibits the expression of these
genes at the transcriptional level.
TSG blocks LPS-induced NF-κB activity
in LPS-stimulated RAW 264.7 macrophages
NF-κB is an important transcription factor that is
required for the activation of pro-inflammatory mediators and
cytokines in LPS-stimulated macrophages. Under normal conditions,
NF-κB is localized to the cytosol due to its binding with IκB.
However, when cells are stimulated by LPS, IκB protein is rapidly
phosphorylated by IκB kinase and degraded, and the NF-κB is
translocated into the nucleus (7).
Therefore, to determine whether TSG prevents the translocation of
NF-κB from the cytosol to the nucleus, western blot analysis was
performed using the nuclear and cytosolic fractions of RAW 264.7
cells. Fig. 6A demonstrates that the
protein expression level of the nuclear p65 subunit of NF-κB was
markedly increased following exposure to LPS alone, indicating that
LPS induced the translocation of NF-κB p65 from the cytosol to the
nucleus. However, the LPS-induced accumulation of NF-κB p65 in the
nuclear fractions was reduced by TSG pretreatment in a
concentration-dependent manner (Fig.
6A). In addition, IκB-α exhibited minor degradation following
exposure to LPS; however, TSG blocked the LPS-induced degradation
of IκB-α in a dose-dependent manner (Fig. 6B). These findings indicate that TSG
inhibits NF-κB activation in RAW 264.7 cells through the
suppression of IκB degradation and the nuclear translocation of
NF-κB.
TSG reduces LPS-induced activation of
MAPKs in LPS-stimulated RAW 264.7 macrophages
As the MAPK signaling pathway is known to be
important for the expression of pro-inflammatory genes, MAPKs act
as specific targets for inflammatory responses (9,10). To
examine whether inhibition of the LPS-induced inflammatory action
by TSG is mediated through MAPK signaling pathways, the effects of
TSG on the activation of MAPKs, including p38 MAPK, ERK and JNK,
were investigated in LPS-stimulated RAW 264.7 cells. As indicated
in Fig. 7, phosphorylation of p38
MAPK, ERK and JNK were induced by stimulation with LPS, whereas TSG
attenuated the LPS-induced phosphorylation of these kinases in a
dose-dependent manner. However, the total protein expression levels
of the unphosphorylated MAPKs were unaffected by LPS and TSG
treatment. These results suggest that TSG may block the LPS-induced
expression of pro-inflammatory responses by inhibiting the MAPK
signaling pathway.
Discussion
The inflammatory process is typically highly
regulated, consisting of signals that initiate and maintain
inflammation, and signals that inhibit the process (4,5). Due to
the importance of activated macrophages in the initiation and
amplification of a variety of inflammatory diseases, therapeutic
methods to reduce the number of activated macrophages, inhibit
their activation signals and/or specific macrophage receptors, or
to selectively counteract macrophage products, are under
investigation for controlling inflammatory diseases (1). In a previous study, it was demonstrated
that TSG was able to exert anti-inflammatory effects in
LPS-activated RAW 264.7 macrophages (19). While this is in agreement with the
results of the present study, the molecular mechanisms have not
been well characterized. The current study also demonstrated that
the anti-inflammatory properties of TSG may be associated with a
reduction in the activation levels of NF-κB and MAPK signaling
pathways.
NO is a highly reactive oxidant that is produced
through the action of iNOS, and participates in diverse biological
mechanisms as a potent pro-inflammatory mediator (21,22).
iNOS is present at low levels under normal physiological
conditions, however, it is rapidly induced by pro-inflammatory and
mitogenic stimuli, including LPS (2,18).
Numerous studies have revealed that excessive NO production is
important in the pathogenesis of inflammation and can lead to
tissue damage by reacting with reactive oxygen species (23,24).
Several inhibitors of NO induction have been reported to exert
anti-inflammatory effects by preventing iNOS expression (17,18,22). In
the present study, TSG was found to significantly reduce the
production of NO in LPS-stimulated RAW 264.7 macrophages by
inhibiting mRNA and protein levels of iNOS, suggesting that the
specific inhibition of the iNOS gene may be responsible for the
anti-inflammatory capacity of TSG. An MTT assay confirmed that the
inhibitory effects of TSG were not a result of cytotoxicity.
Inflammatory disorders are characterized, among
other events, by the production of significant quantities of free
radicals, nitrogen reactive species and cytokines (7,21,23–25).
TNF-α and IL-1β are considered to be critical cytokines in the
inflammatory cytokine network, and are produced as a result of
endotoxin exposure (2–5). Furthermore, TNF-α and IL-1β are
important for promoting the expression of iNOS, in addition to the
production of other cytokines, such as IL-6 and IL-8 (21,25).
Therefore, suppressing the overproduction and activity of
pro-inflammatory cytokines is necessary to reduce inflammation and
its symptoms, and this method has proved to be successful in the
treatment of certain inflammatory diseases, including rheumatoid
arthritis, obesity, diabetes mellitus, cancer and atherosclerosis
(2,5,21,23). In
the present study, it was demonstrated that TSG is able to
significantly suppress TNF-α and IL-1β production. mRNA and protein
expression levels of these cytokines were also significantly
inhibited, suggesting that TSG exerts its anti-inflammatory effects
by suppressing pro-inflammatory cytokines at the transcriptional
level.
As NF-κB and MAPK signaling molecules are key
regulators of a variety of genes involved in inflammatory responses
(8,25), the effects of TSG on the NF-κB and
MAPK signaling pathways were investigated in LPS-induced RAW 264.7
macrophages, in order to further elucidate the underlying
mechanisms of anti-inflammation. Under normal physiological
conditions, NF-κB is localizes in the cytosol as an inactive
heterodimer consisting of three subunits: p50, p65 and Iκ-B. Upon
its activation by various stimuli, including LPS, the Iκ-B protein
is phosphorylated and degraded, resulting in the liberation of
NF-κB and enabling it to translocate to the nucleus (8,25). The
translocation of NF-κB into the nucleus triggers the expression of
target genes, including pro-inflammatory cytokines, adhesion
molecules, chemokines and inducible enzymes, such as iNOS (26,27). The
production of the aforementioned genes acts as a positive autocrine
feedback signal to augment NF-κB activation, and subsequently
increases the production of these pro-inflammatory factors
(7,8,28). The
results of the present study demonstrated that the translocation of
the p65 subunit of NF-κB to the nucleus, in addition to the
degradation of IκB-α, was markedly inhibited by pretreatment with
TSG. Thus, the data indicated that TSG may exert its
anti-inflammatory activity through NF-κB activation; by suppressing
the degradation of IκB-α and the subsequent translocation of NF-κB
p65 from the cytosol to the nucleus in LPS-stimulated RAW 264.7
cells. The present data provided a basis for modeling the effects
of TSG in animal models, in order to facilitate the development of
nutraceutical products and anti-inflammatory drugs based on
TSG.
The MAPK family is a group of serine and threonine
kinases that are activated in response to diverse extracellular
stimuli to mediate signal transduction from the cell surface to the
nucleus (29). Upon the activation
of MAPKs, transcription factors that are present in the cytoplasm
or nucleus are phosphorylated and activated. This activation leads
to the expression of target genes that regulate cell growth,
division, and/or differentiation (30,31). As
described previously, in the LPS-stimulated signaling pathway of
macrophages, MAPKs have demonstrated an association with
LPS-stimulated iNOS and pro-inflammatory cytokine expressions
(9,10). In the present study, it was
identified that the phosphorylation levels of three MAPKs, ERK, JNK
and p38 MAPK, were markedly increased in LPS-stimulated RAW 264.7
cells, compared with unstimulated cells; however, following TSG
administration, the phosphorylation levels of the MAPKs markedly
reduced in a dose-dependent manner. Therefore, it is postulated
that attenuation of the phosphorylation levels of MAPKs by TSG may
contribute to the inhibition of inflammatory reactions in
LPS-stimulated RAW 264.7 cells.
In conclusion, TSG has the ability to inhibit the
production of inflammatory cytokines, in addition to NO, in
LPS-stimulated RAW 264.7 macrophages, by modulating the activation
of intracellular signaling pathways, including NF-κB and multiple
MAPK signaling pathways. Thus, TSG may be a potential candidate for
the treatment of various inflammatory diseases that are associated
with the over-activation of macrophages.
Acknowledgements
The present study was supported by Blue-Bio Industry
Regional Innovation Center at Dongeui University (Busan, Korea), as
an RIC program under the Ministry of Trade, Industry and Energy and
Busan City (RIC; grant no. RIC08-06-07), and the Basic Science
Research Program through a National Research Foundation of Korea
grant funded by the Korea government (grant no.
2015R1A2A2A01004633).
References
|
1
|
Zhang X and Mosser DM: Macrophage
activation by endogenous danger signals. J Pathol. 214:161–178.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fujihara M, Muroi M, Tanamoto K, Suzuki T,
Azuma H and Ikeda H: Molecular mechanisms of macrophage activation
and deactivation by lipopolysaccharide: Roles of the receptor
complex. Pharmacol Ther. 100:171–194. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Park BS and Lee JO: Recognition of
lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med.
45:e662013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kapadia M and Sakic B: Autoimmune and
inflammatory mechanisms of CNS damage. Prog Neurobiol. 95:301–333.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Laveti D, Kumar M, Hemalatha R, Sistla R,
Naidu VG, Talla V, Verma V, Kaur N and Nagpal R: Anti-inflammatory
treatments for chronic diseases: A review. Inflamm Allergy Drug
Targets. 12:349–361. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Muralidharan S and Mandrekar P: Cellular
stress response and innate immune signaling: Integrating pathways
in host defense and inflammation. J Leukoc Biol. 94:1167–1184.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Guha M and Mackman N: LPS induction of
gene expression in human monocytes. Cell Signal. 13:85–94. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li Q and Verma IM: NF-kappaB regulation in
the immune system. Nat Rev Immunol. 2:725–734. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Caivano M: Role of MAP kinase cascades in
inducing arginine transporters and nitric oxide synthetase in
RAW264 macrophages. FEBS Lett. 429:249–253. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Uto T, Fujii M and Hou DX:
6-(Methylsulfinyl)hexyl isothiocyanate suppresses inducible nitric
oxide synthase expression through the inhibition of Janus kinase
2-mediated JNK pathway in lipopolysaccharide-activated murine
macrophages. Biochem Pharmacol. 70:1211–1221. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Choi J, Kim TH, Choi TY and Lee MS:
Ginseng for health care: A systematic review of randomized
controlled trials in Korean literature. PLoS One. 8:e599782013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Coleman CI, Hebert JH and Reddy P: The
effects of Panax ginseng on quality of life. J Clin Pharm
Ther. 28:5–15. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kitts D and Hu C: Efficacy and safety of
ginseng. Public Health Nutr. 3(4A): 473–485. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shibata S: Chemistry and cancer preventing
activities of ginseng saponins and some related triterpenoid
compounds. J Korean Med Sci. 16:S28–37. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen RJ, Chung TY, Li FY, Lin NH and Tzen
JT: Effect of sugar positions in ginsenosides and their inhibitory
potency on Na+/K+-ATPase activity. Acta
Pharmacol Sin. 30:61–69. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lü JM, Yao Q and Chen C: Ginseng
compounds: An update on their molecular mechanisms and medical
applications. Curr Vasc Pharmacol. 7:293–302. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim JY, Lee HJ, Kim JS and Ryu JH:
Induction of nitric oxide synthase by saponins of heat-processed
ginseng. Biosci Biotechnol Biochem. 69:891–895. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Park JS, Park EM, Kim DH, Jung K, Jung JS,
Lee EJ, Hyun JW, Kang JL and Kim HS: Anti-inflammatory mechanism of
ginseng saponins in activated microglia. J Neuroimmunol. 209:40–49.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kang A, Hao H, Zheng X, Liang Y, Xie Y,
Xie T, Dai C, Zhao Q, Wu X, Xie L and Wang G: Peripheral
anti-inflammatory effects explain the ginsenosides paradox between
poor brain distribution and anti-depression efficacy. J
Neuroinflammation. 8:1002011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sugimoto S, Nakamura S, Matsuda H,
Kitagawa N and Yoshikawa M: Chemical constituents from seeds of
Panax ginseng: Structure of new dammarane-type triterpene
ketone, panaxadione, and hplc comparisons of seeds and flesh. Chem
Pharm Bull (Tokyo). 57:283–287. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dinarello CA: Proinflammatory cytokines.
Chest. 118:503–508. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Koppula S, Kumar H, Kim IS and Choi DK:
Reactive oxygen species and inhibitors of inflammatory enzymes,
NADPH oxidase, and iNOS in experimental models of Parkinson's
disease. Mediators Inflamm. 823902:20122012.
|
|
23
|
Martinon F: Signaling by ROS drives
inflammasome activation. Eur J Immunol. 40:616–619. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Korbecki J, Baranowska-Bosiacka I,
Gutowska I and Chlubek D: The effect of reactive oxygen species on
the synthesis of prostanoids from arachidonic acid. J Physiol
Pharmacol. 64:409–421. 2013.PubMed/NCBI
|
|
25
|
Marcus JS, Karackattu SL, Fleegal MA and
Sumners C: Cytokine-stimulated inducible nitric oxide synthase
expression in astroglia: role of Erk mitogen-activated protein
kinase and NF-kappaB. Glia. 41:152–160. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mercurio F and Manning AM: NF-kappaB as a
primary regulator of the stress response. Oncogene. 18:6163–6171.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Klemm S and Ruland J: Inflammatory signal
transduction from the Fc epsilon RI to NF-kappa B. Immunobiology.
211:815–820. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sonis ST: The biologic role for nuclear
factor-kappaB in disease and its potential involvement in mucosal
injury associated with anti-neoplastic therapy. Crit Rev Oral Biol
Med. 13:380–389. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim EK and Choi EJ: Pathological roles of
MAPK signaling pathways in human diseases. Biochim Biophys Acta.
1802:396–405. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huang P, Han J and Hui L: MAPK signaling
in inflammation-associated cancer development. Protein Cell.
1:218–226. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kaminska B, Gozdz A, Zawadzka M,
Ellert-Miklaszewska A and Lipko M: MAPK signal transduction
underlying brain inflammation and gliosis as therapeutic target.
Anat Rec (Hoboken). 292:1902–1913. 2009. View Article : Google Scholar : PubMed/NCBI
|