Introduction
Global cerebral ischemia (GCI), one of the
consequences of surgical procedure and cardiac arrest, is a leading
cause of disability and the second leading cause of mortality
worldwide (1). Previous studies have
revealed that intracellular calcium overload, oxidative stress, and
post-ischemic glutamate and inflammatory cytokine release may be
involved in the pathological process of GCI-induced brain injury
(2–5). A complex interplay between various
factors and signaling cascades leads to neuronal cell injury and
death following ischemia (6,7). Although significant progress has been,
made with extensive animal research into GCI treatment such as
calcium channel blockers, radical scavengers, glutamate receptor
antagonists and anti-inflammatory agents (7), few of these have been translated into
clinically effective therapies (8).
As the most abundant cell type in the brain,
astrocytes represent an attractive cellular candidate for GCI
treatment (9). During the
pathological process of GCI, astrocytes are able to protect neurons
from injury via housekeeping mechanisms (9). Conversely, astrocytes are able to
aggravate brain injury by releasing pro-inflammatory molecules and
glutamate, thereby exacerbating the formation of brain edema
(10,11). However, few studies have investigated
the effect of directly targeting astrocytes in the setting of GCI
(10).
Rosiglitazone (RSG) is a peroxisome proliferating
activating receptor-γ (PPAR-γ) agonist known for its
anti-inflammatory effects (12).
Previous studies have demonstrated that treatment with RSG can
exert neuroprotection in animal models of a number of conditions,
including Alzheimer's disease, traumatic brain injury, spinal cord
injury, and ischemia stroke (13–16). In
addition, a recent study suggested that administration of RSG
provided beneficial effects in the hippocampus of the rat brain
following GCI (17); however,
whether RSG treatment is involved in the astrocyte over-activation
and inflammatory reaction in the cortex remains to be elucidated.
Therefore, the present study aimed to investigate whether RSG
treatment was able to improve functional impairment induced
following GCI and protect against cortex neuron loss, in addition
to elucidating the potential mechanisms underlying these
functions.
Materials and methods
Animals and GCI model
A total of 180 rats adult female Sprague-Dawley rats
(Wuhan University Animal Center, Wuhan, China), weighing 250–300 g
and aged 3 months, were used in the present study. All procedures
were approved by the Animal Care Welfare Committee of Zhongnan
Hospital, Wuhan University for ethical experimentation on animals.
All rats were provided with ad libitum access to food and
water prior to the surgical procedure under optimal conditions
(12-h light/dark cycle, 22°C). Female rats were bilaterally
ovariectomized, and 1 week later, GCI was induced by 4-vessel
occlusion as described previously (18). Briefly, the rats were anesthetized
with 10% chloral hydrate (350 mg/kg, intraperitoneally; Beijing
Solarbio Science & Technology Co., Ltd., Beijing, China), the
vertebral arteries were electrocauterized and the common carotid
arteries (CCA) were exposed. Following 24 h, the rats were
anesthetized using 0.6 ml/kg isoflurane (intraperitoneally; Beijing
Solarbio Science & Technology Co., Ltd.) and the CCA were
re-exposed and clipped using artery clips for 10 min followed by
reperfusion. Rats that lost their righting reflex within 30 sec,
those that had dilated pupils, and those that lost response to
light during ischemia were selected for the experiments. Rectal
temperature was maintained at 37±0.5°C using a thermal blanket
during ischemia. Sham-operated animals underwent the same surgical
procedures without occlusion of the CCA.
Group and drug administration
A total of 180 rats were randomly assigned to three
groups: Sham-operated group (Sham, n=60); GCI group that received
only equal volumes of 0.9% saline solution (GCI, n=60); and a group
treated with 2 mg/kg RSG (Cell Signaling Technology, Inc., Danvers,
MA, USA) following GCI (RSG, n=60). RSG was dissolved in 0.9%
saline and stored at 4°C. Following GCI, RSG was immediately
injected intraperitoneally in the rats of the RSG group following
GCI (2 mg/kg). All tests were blinded, and the animal codes were
revealed only at the end of the behavioral and histological
analyses.
Measurement of the neurological
deficit and infarct volume
Neurological deficit was evaluated 24 h following
reperfusion according to the method described by Longa et al
(18). Rats were anesthetized with
50 mg/kg sodium pentobarbital (i.p; Beijing Solarbio Science &
Technology Co., Ltd.) prior to sacrifice via exsanguination. The
brains of the rats were then dissected and sectioned into five 2-mm
coronal sections, that were incubated in 2%
2,3,5-triphenyltetrazolium chloride (TTC; Amresco, LLC, Solon, OH,
USA) for 15 min at 37°C, as previously described (19) with minor modifications. The tissue
sections were subsequently immersed and fixed in 4%
paraformaldehyde (Beijing Solarbio Science & Technology Co.,
Ltd.). The images of TTC-stained tissue sections were captured
using an Olympus FE4000 digital camera (Olympus Corporation, Tokyo,
Japan), and the digital images were analyzed using ImageJ image
analysis software (version 1.41; National Institutes of Health,
Bethesda, MA, USA). Infarct areas were measured and then compiled
to obtain the infarct volume (mm3) for each brain tissue
sample.
Immunofluorescence
Coronal sections were incubated with 10% normal
donkey serum for 30 min at room temperature in phosphate-buffered
saline (PBS) and 0.1% Triton X-100 (all Beijing Solarbio Science
& Technology Co., Ltd.) followed by incubation with appropriate
primary antibodies overnight at 4°C in the same buffer. The frozen
tissue sections were incubated with mouse anti-neuron-specific
nuclear protein (NeuN) polyclonal antibody (1:100; sc-31154) and
mouse anti-glial fibrillary acidic protein (GFAP) monoclonal
immunoglobulin (Ig)G1 (F7) antibody (1:100; sc-166458;
both Santa Cruz Biotechnology, Inc., Dallas, TX, USA) overnight at
4°C. The following day, the tissue sections were incubated with
mouse FITC monoclonal IgG1 (1:1,000; sc-69871; Santa
Cruz Biotechnology, Inc.) for 2 h at 37°C in the dark. Images were
captured using a laser scanning confocal microscope (Olympus
FV1000; Olympus Corporation, Tokyo, Japan). Primary antibodies were
replaced with PBS in the negative control group.
Western blot analysis
Western blot analysis was conducted according to
standard protocols (20). Rats were
anesthetized using 50 mg/kg sodium pentobarbital and were
intracardically perfused with 0.1 mol/l PBS (pH 7.4). The cortex
region of the brain was rapidly isolated, homogenized (BestBio
Biotechnology, Beijing, China), and total proteins were extracted
using protein extraction reagent (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA), according to manufacturer's protocol. Protein
concentration was determined using a bicinchoninic acid assay
(Beijing Solarbio Science & Technology Co., Ltd.). Briefly, 35
µg total protein was separated by 20% SDS-polyacrylamide gel
electrophoresis and electroblotted onto polyvinylidene fluoride
membranes (EMD Millipore, Billerica, MA, USA) prior to being
blocking with 5% fat-free dry milk for 1 h at room temperature. The
membranes were subsequently incubated with the following primary
antibodies overnight at 4°C: Rabbit anti-interleukin (IL)-1β
polyclonal antibody (1:500; sc-7884), rabbit anti-IL-6 polyclonal
antibody (1:500; sc-7920), rabbit anti-glutamate transporter
(GLT)-1 polyclonal antibody (1:500; sc-15317), rabbit anti-tumor
necrosis factor-α (TNF-α) polyclonal antibody (1:500; sc-7895),
mouse anti-β-actin polyclonal antibody (1:500; sc-376421; all Santa
Cruz Biotechnology, Inc.). Membranes were subsequently washed twice
with Tris-buffered saline with Tween-20 (TBST) for 20 min and prior
to incubation with horseradish peroxidase-conjugated anti-rabbit
IgG (1:5,000; sc-2027) and anti-mouse IgG (1:5,000; sc-2025; both
Santa Cruz Biotechnology, Inc.) for 2 h at room temperature.
Membranes were washed four times with TBST for 40 min. Protein
bands on the membrane were visualized using an enhanced
chemiluminescent reagent (EMD Millipore) and densitometric signals
were quantified using ImageJ software (version 1.41; National
Institutes of Health).
Statistical analysis
All data were expressed as the mean ± standard error
of the mean. One way analysis of variance was used to assess
statistical differences among the groups using SPSS 17.0 software
(SPSS, Inc., Chicago, IL, USA). Significant differences between
groups at each time point were assessed by Student's t-test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Treatment with RSG attenuates
GCI-induced neurological deficits
Fig. 1 shows the
changes in neurological deficit scores in the three groups.
Post-injury administration of RSG significantly (P<0.05)
improved neurological function recovery.
Treatment with RSG attenuates cerebral
infarct volume
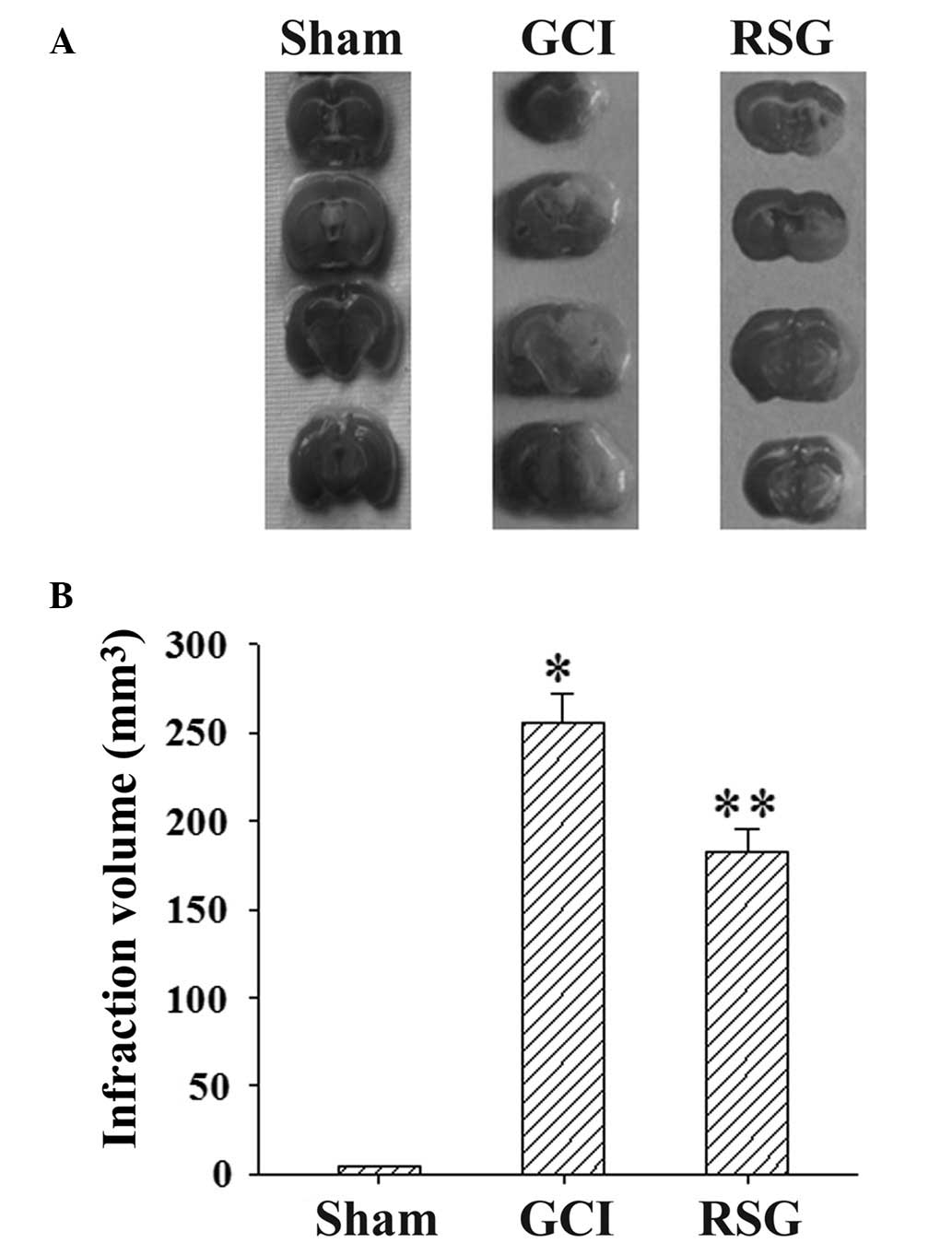
Ischemia/reperfusion produced marked infarction, as
demonstrated in the serial coronal brain tissue sections (Fig. 2A). RSG treatment significantly
(P<0.05) reduced the infarct volume, as compared with the GCI
group at 24 h post-GCI (Fig.
2B).
Treatment with RSG suppresses neuronal
death in the cortex following GCI
The neuronal survival rate in the rat cortex was
assessed using anti-NeuN antibody at 24 h post-GCI. As shown in
Fig. 3, GCI caused significant
(P<0.01) neuron loss compared with the Sham group; however, this
effect was partly reversed by treatment with RSG.
Treatment with RSG attenuates
astrocyte over-activation induced by GCI
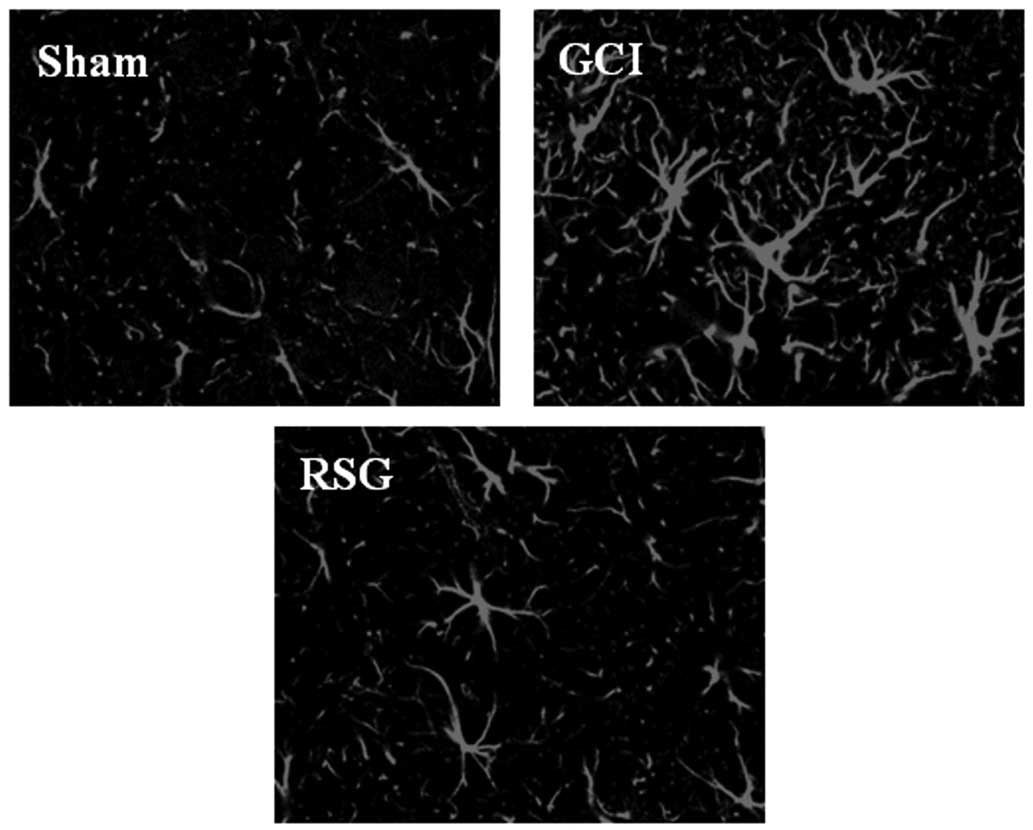
It was next examined whether RSG treatment affected
astrocytes in the rat cortex by using anti-GFAP antibodies as a
marker for activated astrocytes. As shown in Fig. 4, GCI induced marked astrocyte
over-activation. However, RSG caused a marked attenuation of
astrocyte activation compared with the GCI group.
Treatment with RSG attenuates IL-1β
expression in the rat cortex following GCI
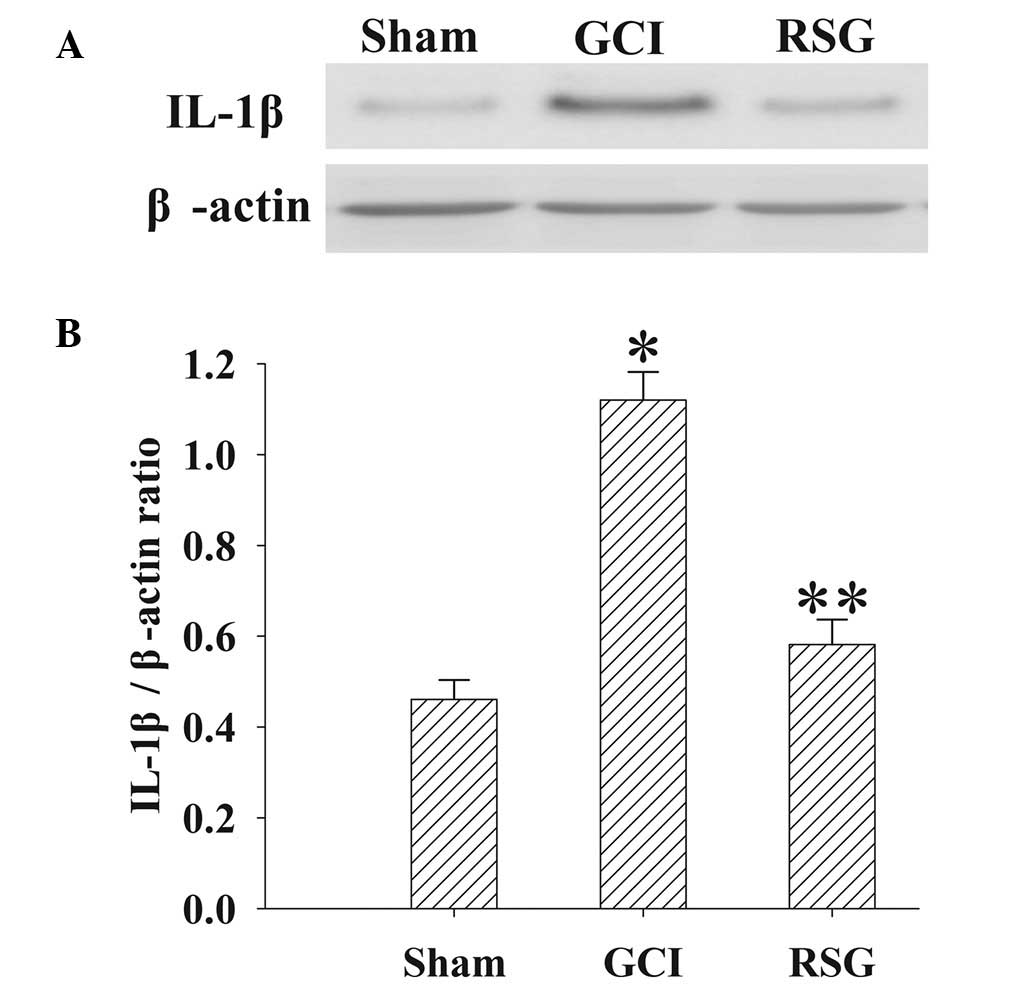
The expression levels of IL-1β in the cortex at 24 h
were measured by western blotting (Fig.
5). IL-1β expression levels were significantly elevated in the
GCI group, as compared with the Sham group. However, treatment with
RSG induced a significant (P<0.01) reduction in IL-1β expression
levels.
Treatment with RSG attenuates IL-6
expression levels in the cortex following GCI
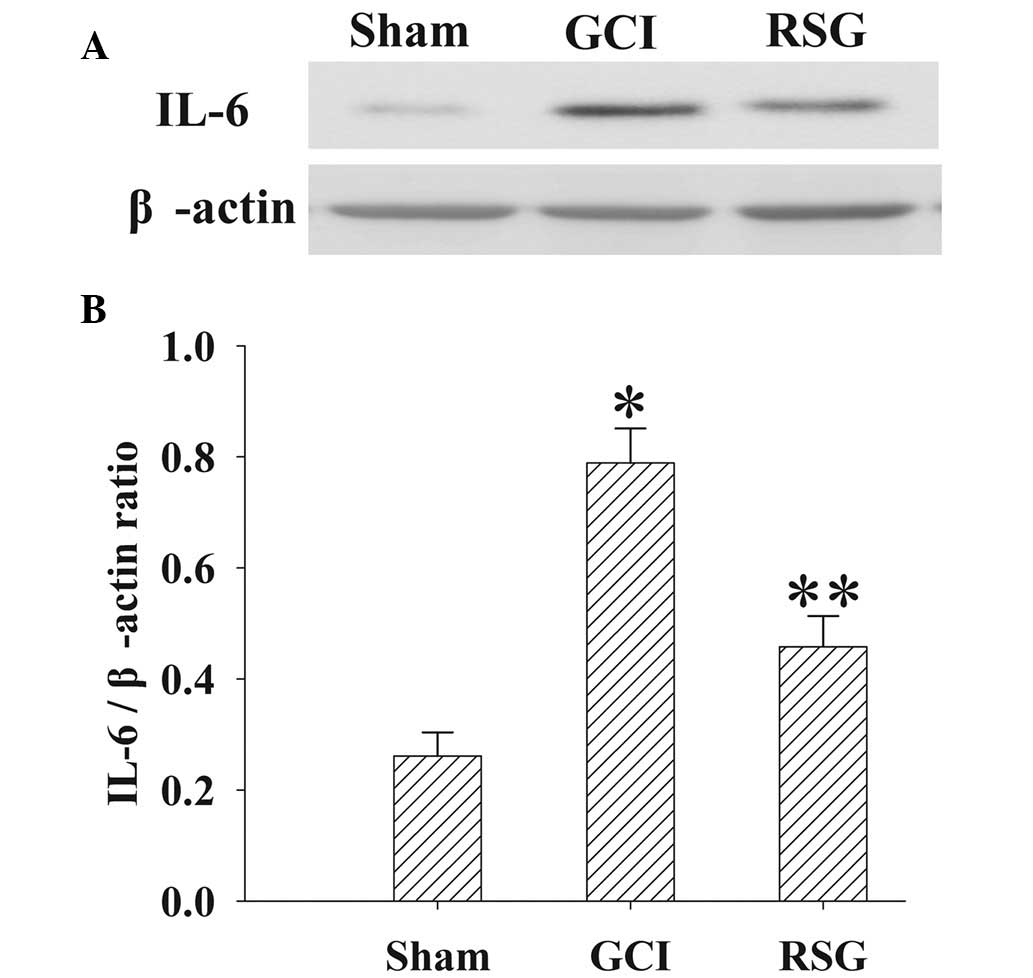
The expression levels of IL-6 in the cortex at 24 h
were measured by western blotting (Fig.
6). IL-6 expression levels were significantly increased
(P<0.01) in the GCI group, as compared with the Sham group.
However, treatment with RSG significantly (P<0.01) decreased
IL-6 expression levels, as compared with the GCI group.
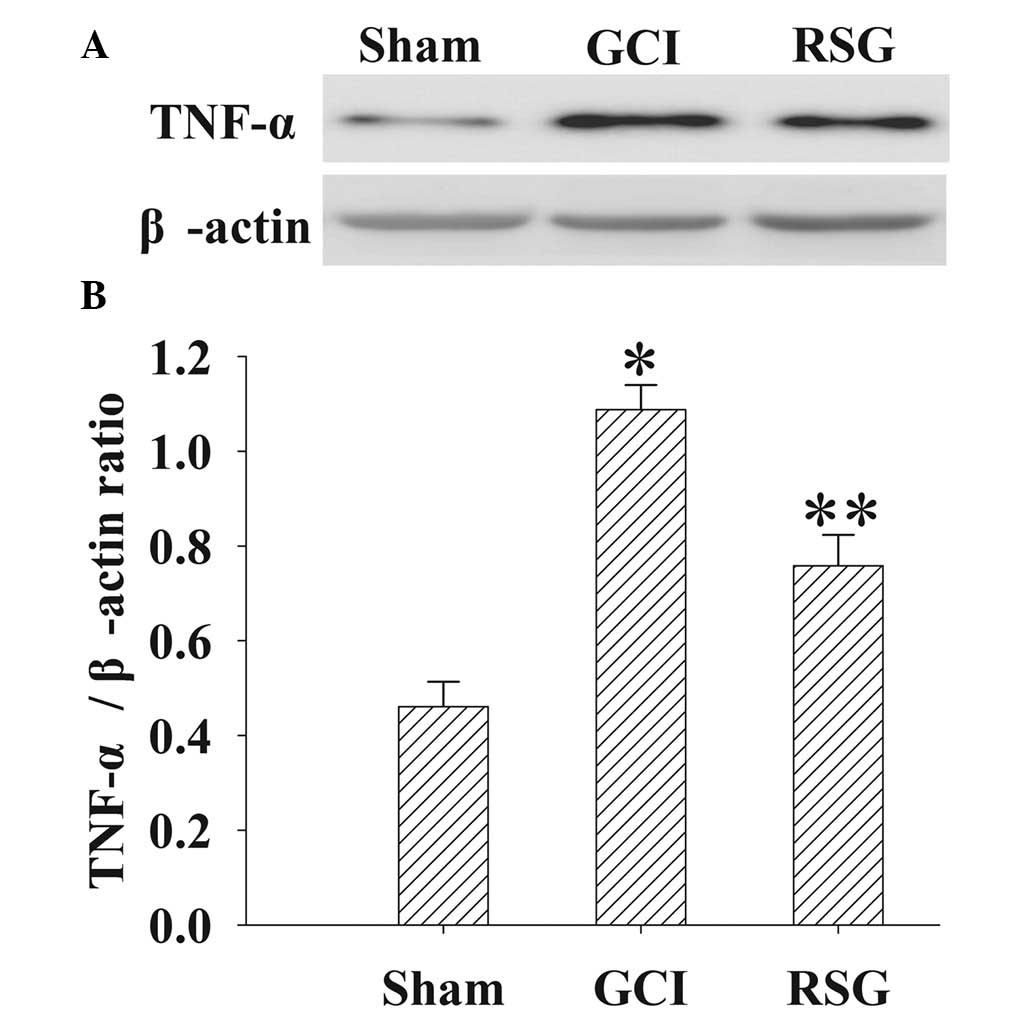
Treatment with RSG attenuates TNF-α
expression levels in the cortex following GCI
The expression levels of TNF-α in the cortex at 24 h
were measured by western blotting (Fig.
7). TNF-α expression levels were significantly (P<0.01)
elevated in the GCI group, as compared with the Sham group.
However, administration of RSG induced a significant (P<0.01)
reduction in the GCI-induced upregulation of TNF-α expression
levels.
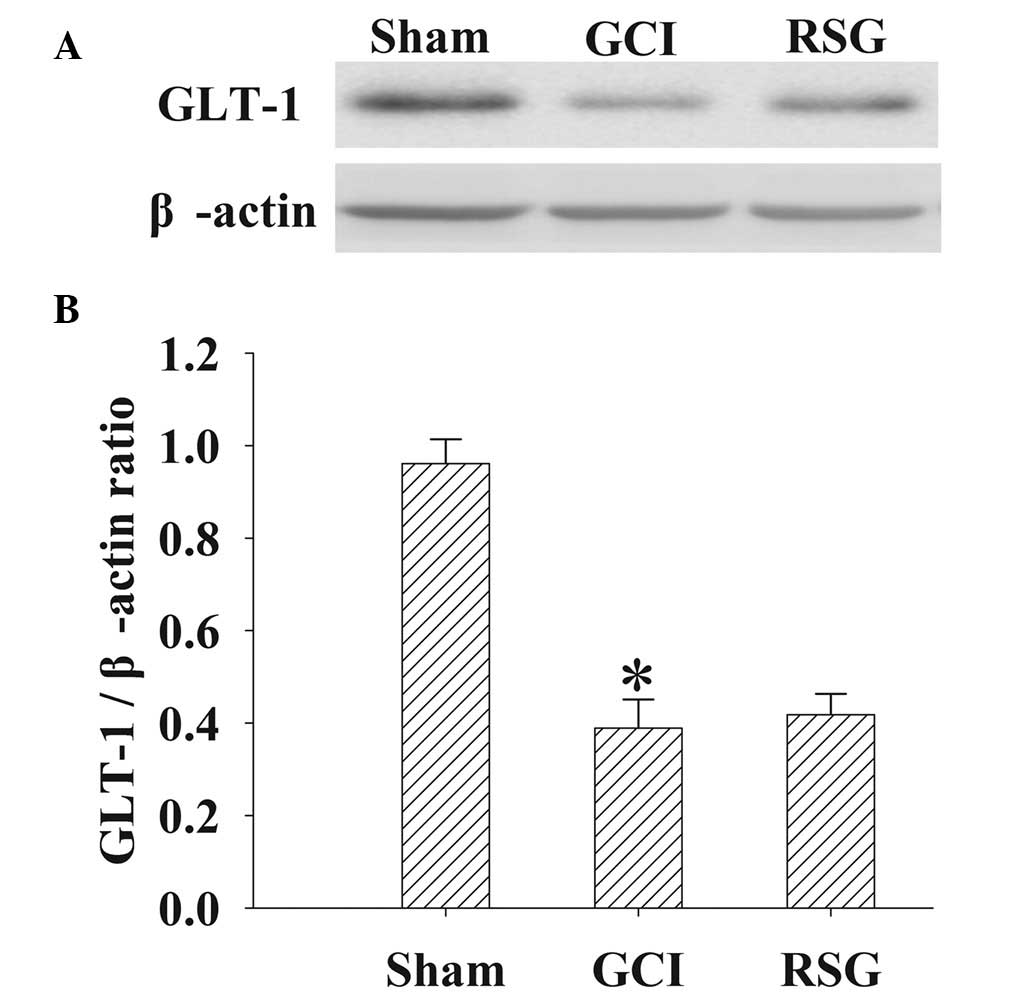
No significant changes in GLT-1
expression levels were observed in the cortex tissue samples
following RSG treatment
GLT-1 protein expression levels in the cortex were
analyzed by western blot analysis at 24 h (Fig. 8). Significant (P<0.01)
downregulation in GLT-1 expression levels was detected in the GCI
group, as compared with the Sham group. However, treatment with RSG
induced no significant (P>0.05) changes in GLT-1 expression
levels.
Discussion
GCI is a leading cause of mortality worldwide and
remains the primary cause of long-term neurological disability
(21). Astrocyte over-activation as
well as extensive loss of neurons in the ischemic brain are the
characteristic pathological features of ischemia stroke (22). One reason for the failure to
translate successful results in animal experiments to clinical
therapies may be due to the complexity of signaling responses which
reduce the likelihood that altering a single target will be
effective (6,7). The present study investigated the
efficacy of the PPAR-γ agonist, RSG, as a therapeutic strategy for
the treatment of GCI. The results demonstrated that RSG treatment
immediately following GCI significantly reduced infarct volume and
neuron survival rate, in addition to increasing functional
recovery. Furthermore, these results correlated with a reduction in
astrocyte over-activation and inflammatory cytokines in the rat
cortex. Previous studies have demonstrated that RSG provides
neuroprotective effects in numerous acute and chronic brain injury
models, including amyotrophic lateral sclerosis, Alzheimer's
disease, traumatic brain injury, spinal cord injury and ischemia
stroke (13–16). The data of the present study were
concordant with those of previous investigations, and to the best
of our knowledge reported for the first time that post-GCI
administration of RSG provided neuroprotective effects via
attenuation of astrocyte over-activation in the cortex.
An important delayed mechanism beginning within
hours of the onset of GCI-induced brain injury is the inflammatory
response in the ischemic tissue (22). In particular, cerebral ischemia
rapidly elevated inflammatory responses in the rat brain, thereby
contributing to blood brain barrier disruption and delayed neuronal
death (5,22). Therefore, therapeutic strategies
targeting the delayed inflammatory response may inhibit the
progression of the tissue damage, which would provide an extended
therapeutic window for neuroprotection on GCI. However, during the
response to ischemic injury, microglia and astrocytes are activated
in the brain (23). Astrocytes are
sensitive to the increased release of these immunomodulatory
peptides and therefore severe ischemia also compromises astrocytic
function (23,24). Following GCI, astrocytes rapidly
become over-activated and undergo morphological transformations,
accompanied by functional changes such as increasing expression
levels of cytokines, including TNF-α, interleukins (IL-1β, IL-4,
IL-6, IL-10), chemokines and interferons (25). Subsequently, the accumulation of
pro-inflammatory factors further exacerbates ischemic damage
(26–28). In the present study, the result
demonstrated that ischemic injury resulted in over-activation of
astrocytes, and thereby significantly elevated the expression
levels of IL-1β, IL-6, and TNF-α in the cortex. Furthermore, it is
worth noting that treatment with RSG was able to inhibit astrocyte
over-activation, and reduce the levels of these inflammatory
factors. Although the role of anti-inflammatory factors in stroke
patients remains to be fully elucidated, the majority of the
existing studies have demonstrated that RSG is able to exert
neuroprotection via its anti-inflammatory activity in numerous
animal models of neurological disorders (13–16).
Therefore, the results herein suggest that RSG exerts its
neuroprotective effects by inhibiting astrocyte over-activation,
and thereby attenuating inflammatory cytokine release.
Astrocytes protect against glutamate excitotoxicity
via glutamate transporters, and GLT-1 is responsible for ~90% of
all glutamate transport in adult brain tissue (29). Previous studies have demonstrated
that Ceftriaxone treatment, which induces astrocytic glutamate
uptake via elevated GLT-1, exerted neuroprotection effects in
numerous models of neurological disease (30–32).
Similarly, selective overexpression of GLT-1 in astrocytes also
provided neuroprotection following focal or global cerebral
ischemia (33). However, in the
present study, no significant changes in GLT-1 expression levels
were observed following RSG treatment compared with the GCI rats,
which suggested that the neuroprotective effect of RSG does not
appear to be mediated by the modulation of GLT-1 protein expression
levels in the rat model of GCI.
In conclusion, the present investigation
demonstrated that RSG significantly protected rats against
ischemia-reperfusion-induced brain injury. In addition, RSG may
exert neuroprotective effects by inhibiting astrocyte
over-activation, and thereby reduces the levels of inflammatory
cytokines in the GCI-injured brain.
Acknowledgements
The present study was supported by a grant from the
Nature Science Foundation of Hubei Province (grant no.
2014CFB479).
Glossary
Abbreviations
Abbreviations:
|
RSG
|
rosiglitazone
|
|
GCI
|
global cerebral ischemia
|
|
NeuN
|
neuron-specific nuclear protein
|
|
GFAP
|
glial fibrillary acidic protein
|
|
TNF-α
|
tumor necrosis factor α
|
|
IL-1β
|
interleukin-1β
|
|
IL-6
|
interleukin-6
|
References
|
1
|
Cao Y, Mao X, Sun C, Zheng P, Gao J, Wang
X, Min D, Sun H, Xie N and Cai J: Baicalin attenuates global
cerebral ischemia/reperfusion injury in gerbils via anti-oxidative
and anti-apoptotic pathways. Brain Res Bull. 85:396–402. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kristián T and Siesjö BK: Calcium in
ischemic cell death. Stroke. 29:705–718. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Love S: Oxidative stress in brain
ischemia. Brain Pathol. 9:119–131. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Choi DW and Rothman SM: The role of
glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu
Rev Neurosci. 13:171–182. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huang J, Upadhyay UM and Tamargo RJ:
Inflammation in stroke and focal cerebral ischemia. Surg Neurol.
66:232–245. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shamloo M, Rytter A and Wieloch T:
Activation of the extracellular signal-regulated protein kinase
cascade in the hippocampal CA1 region in a rat model of global
cerebral ischemic preconditioning. Neuroscience. 93:81–88. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mehta SL, Manhas N and Raghubir R:
Molecular targets in cerebral ischemia for developing novel
therapeutics. Brain Res Rev. 54:34–66. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu S, Levine SR and Winn HR: Targeting
ischemic penumbra: Part I-from pathophysiology to therapeutic
strategy. J Exp Stroke Transl Med. 3:47–55. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ogata K and Kosaka T: Structural and
quantitative analysis of astrocytes in the mouse hippocampus.
Neuroscience. 113:221–233. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pekny M and Nilsson M: Astrocyte
activation and reactive gliosis. Glia. 50:427–434. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Swanson RA, Ying W and Kauppinen TM:
Astrocyte influences on ischemic neuronal death. Curr Mol Med.
4:193–205. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mohanty P, Aljada A, Ghanim H, Hofmeyer D,
Tripathy D, Syed T, Al-Haddad W, Dhindsa S and Dandona P: Evidence
for a potent antiinflammatory effect of rosiglitazone. J Clin
Endocrinol Metab. 89:2728–2735. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Escribano L, Simón A-M, Pérez-Mediavilla
A, Salazar-Colocho P, Del Río J and Frechilla D: Rosiglitazone
reverses memory decline and hippocampal glucocorticoid receptor
down-regulation in an Alzheimer's disease mouse model. Biochem
Biophys Res Commun. 379:406–410. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yi JH, Park SW, Brooks N, Lang BT and
Vemuganti R: PPARgamma agonist rosiglitazone is neuroprotective
after traumatic brain injury via anti-inflammatory and
anti-oxidative mechanisms. Brain Res. 1244:164–172. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Q, Hu W, Meng B and Tang T: PPAR γ
agonist rosiglitazone is neuroprotective after traumatic spinal
cord injury via anti-inflammatory in adult rats. Neurol Res.
32:852–859. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Luo Y, Yin W, Signore AP, Zhang F, Hong Z,
Wang S, Graham SH and Chen J: Neuroprotection against focal
ischemic brain injury by the peroxisome proliferator-activated
receptor-gamma agonist rosiglitazone. J Neurochem. 97:435–448.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Al Rouq F and El Eter E: PPAR-γ activator
induces neuroprotection in hypercholesterolemic rats subjected to
global cerebral ischemia/reperfusion injury: In vivo and in vitro
inhibition of oxidative stress. Exp Gerontol. 51:1–7. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bederson JB, Pitts LH, Germano SM,
Nishimura MC, Davis RL and Bartkowski HM: Evaluation of
2,3,5-triphenyltetrazolium chloride as a stain for detection and
quantification of experimental cerebral infarction in rats. Stroke.
17:1304–1308. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shimamura N, Matchett G, Solaroglu I,
Tsubokawa T, Ohkuma H and Zhang J: Inhibition of integrin αvbeta3
reduces blood-brain barrier breakdown in focal ischemia in rats. J
Neurosci Res. 84:1837–1847. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Strong K, Mathers C and Bonita R:
Preventing stroke: Saving lives around the world. Lancet Neurol.
6:182–187. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vexler ZS, Tang XN and Yenari MA:
Inflammation in adult and neonatal stroke. Clin Neurosci Res.
6:293–313. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Willis CL: Glia-induced reversible
disruption of blood-brain barrier integrity and neuropathological
response of the neurovascular unit. Toxicol Pathol. 39:172–185.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang Q, Tang XN and Yenari MA: The
inflammatory response in stroke. J Neuroimmunol. 184:53–68. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pickering M and O'Connor JJ:
Pro-inflammatory cytokines and their effects in the dentate gyrus.
Prog Brain Res. 163:339–354. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kaushal V and Schlichter LC: Mechanisms of
microglia-mediated neurotoxicity in a new model of the stroke
penumbra. J Neurosci. 28:2221–2230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Barone FC and Parsons AA: Therapeutic
potential of anti-inflammatory drugs in focal stroke. Expert Opin
Investig Drugs. 9:2281–2306. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Batti L and O'Connor JJ: Tumor necrosis
factor-alpha impairs the recovery of synaptic transmission from
hypoxia in rat hippocampal slices. J Neuroimmunol. 218:21–27. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Anderson CM and Swanson RA: Astrocyte
glutamate transport: Review of properties, regulation and
physiological functions. Glia. 32:1–14. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chu K, Lee ST, Sinn DI, Ko SY, Kim EH, Kim
JM, Kim SJ, Park DK, Jung KH, Song EC, et al: Pharmacological
Induction of ischemic tolerance by glutamate transporter-1 (EAAT2)
upregulation. Stroke. 38:177–182. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wei J, Pan X, Pei Z, Wang W, Qiu W, Shi Z
and Xiao G: The beta-lactam antibiotic, ceftriaxone, provides
neuroprotective potential via anti-excitotoxicity and
anti-inflammation response in a rat model of traumatic brain
injury. J Trauma Acute Care Surg. 73:654–660. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Azbill RD, Mu X and Springer JE: Riluzole
increases high-affinity glutamate uptake in rat spinal cord
synaptosomes. Brain Res. 871:175–180. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Harvey BK, Airavaara M, Hinzman J, Wires
EM, Chiocco MJ, Howard DB, Shen H, Gerhardt G, Hoffer BJ and Wang
Y: Targeted over-expression of glutamate transporter 1 (GLT-1)
reduces ischemic brain injury in a rat model of stroke. PLoS One.
6:e221352011. View Article : Google Scholar : PubMed/NCBI
|