Introduction
Pulmonary alveolar microlithiasis (PAM) is a rare
genetic diffuse lung disease characterized by calcifications within
the alveolar airspaces. It is caused by inactivating mutations in
the solute carrier family 34, member 2 (SLC34A2) gene, which
encodes the type IIb sodium phosphate cotransporter in alveolar
type II cells (1–3). SLC34A2 is primarily expressed in
alveolar type II cells, where it is responsible for the transport
of phosphate ions from the alveolar space into the alveolar type II
cells (4). Therefore, the inability
of the alveolar type II cells to remove phosphorus ions from the
alveolar space as a result of inactivating mutations in SLC34A2,
leads to microlith formation in the alveolar space (2,4).
PAM has been reported in all continents, with no
particular geographic or racial distribution. However, a review of
1,022 published cases showed that PAM was more prevalent in Asia,
followed by Europe, North America, South America and Africa, with
the majority of cases reported in Turkey, followed by China, Japan,
India, Italy and the USA (1).
Furthermore, the ratio between the number of cases per 1 million
people was 1.85 for Turkey, 1.08 for Italy, 0.92 for Japan, 0.15
for the USA, 0.1 for China and 0.06 for India (1). PAM may affect people of any age,
ranging from premature infants to the elderly; the youngest
reported case was of premature twins (5), and the eldest patient to be diagnosed
with PAM was an 84-year-old female (6). Familial occurrence has been observed in
50% of Japanese, 48% of Turkish and 43.7% of Italian patients
(7). A recent review analyzing 1,022
patients worldwide showed that 37.2% (381 of 1022 patients,
belonging to 163 families) of PAM cases have familial presentation
(1). Furthermore, in 36 of 163
families, the parents were cousins (1).
The hallmark of PAM is the striking dissociation
between the radiological appearance and clinical presentation,
meaning that a patient may present with a paucity of symptoms in
contrast to image findings (2).
Frequently, patients may have no clinical symptoms, such that
diagnosis is often fortuitous (8).
In symptomatic patients, dyspnoea is the most frequently
encountered symptom, followed by a cough, chest pain and asthenia
(8). The radiological appearance on
high resolution computed tomography (HRCT) scans include
ground-glass opacities, small parenchymal nodules, small subpleural
nodules, subpleural cysts and subpleural linear calcifications
(9). Given the striking dissociation
between the radiological appearance and clinical presentation of
PAM, the diagnosis may be based on typical radiological findings,
particularly in family members of a patient previously diagnosed
with PAM (2).
At present, no definitive treatment is available to
reduce the disease progression. Systemic corticosteroids,
calcium-chelating agents and serial bronchopulmonary lavage have
been shown to be ineffective and are used as palliative treatments
(1). Disodium etidronate (DE) has
been proposed as an effective medicine to reduce calcium phosphate
precipitation in PAM (10–12).
In the present study, a case of PAM in a 35-year-old
female patient with severe typical imaging findings, mild clinical
manifestation and characteristic histopathology is reported. In
addition, the present study reviewed the typical radiological
appearance, clinical presentation, pathological characteristics and
novel treatments for PAM, and supported that often the parents of
the patients are cousins. Furthermore, the present study proposes
that a bronchoalveolar lavage fluid (BALF) examination may be
considered an alternative method for the pathological diagnosis of
the PAM.
Case report
A 35-year-old female patient presented at the Taihe
Hospital (Shiyan, China) in October 2013 with a 4-year history of a
persistent dry cough. The parents of the patient were reported to
be blood-related (cousins). A physical examination revealed
striking bibasilar inspiratory crackles (velcro rales), and finger
clubbing was observed. An arterial blood gas test revealed that the
oxygen saturation was 96% (normal, ≥95%) and the oxygen partial
pressure was 84 mmHg (normal, ≥80 mmHg) on ambient air. A pulmonary
function examination showed typical features of a restrictive
ventilatory defect with a reduced vital capacity of 55% and a
diffusion defect with a reduced lung transfer factor for carbon
monoxide of 47% of the predicted values (normal, ≥80% predicted
value). The results of a 6-min walk distance test indicated a
reduced exercise capacity [distance walked, 331 m; normal for women
= (2.11 × height in cm) - (2.29 × weight in kg) - (5.78 × age) +
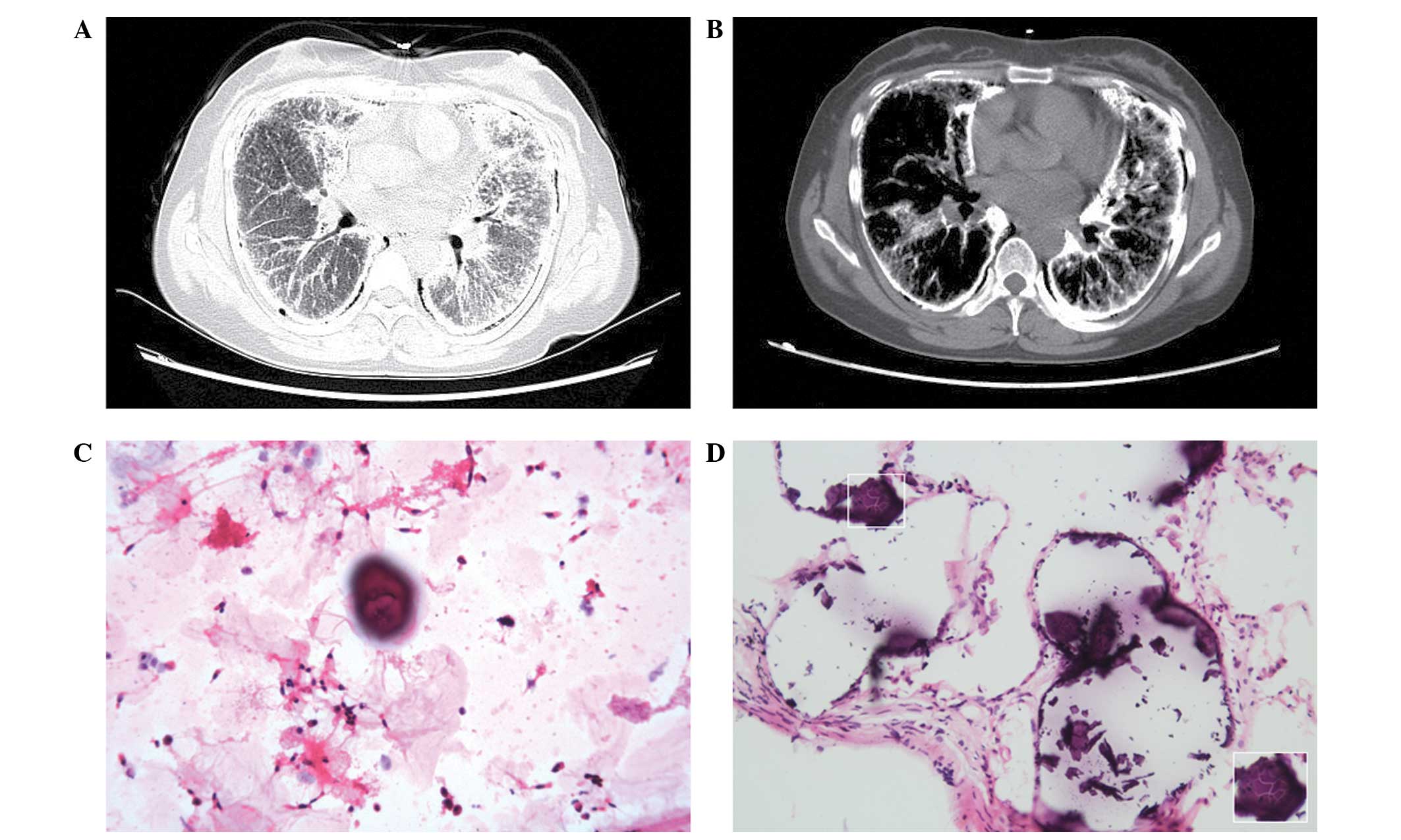
667 m] (13). In addition, a HRCT
scan showed multiple bilateral diffuse ground-glass opacifications
and subpleural linear calcifications. A number of small subpleural
air cysts were observed between the bony rib cage and the calcified
pulmonary infiltrate (also termed the black pleural line).
Subpleural linear calcification and interlobar fissure
calcification were also evident (Fig. 1A
and B).
A BALF examination detected a calcified body in the
BALF and a transbronchial biopsy was subsequently performed. The
lung tissue specimens were fixed in 10% neutral-buffered formalin
(Jiangxi Liansheng Experiment Technic Assembly Co., Ltd., Nanchang,
China) over night, dehydrated through a graded alcohol series,
cleaned with dimethylbenzene and embedded in paraffin (both
Sinopharm Chemical Reagent Co., Ltd., Shanghai, China).
Subsequently, the paraffin-embedded tissue specimens were cut into
5-µm sections, stained with hematoxylin (Shanghai Chemical Reagent
Co., Ltd., Shanghai, China) and eosin (Tianjin Bodi Chemical Co.,
Ltd., Tianjin, China) and then visualized and photographed under a
microscope (BX51; Olympus Corporation, Tokyo, Japan). Numerous
calcified bodies, concentrically laminated with an onion skin-like
appearance in the alveolar spaces, were observed under the
microscope (Fig. 1C and D;
hematoxylin and eosin stain).
Based on the aforementioned observations, the
patient was diagnosed with PAM. Following inhalation of budesonide
suspension (1 mg; AstraZeneca, London, UK), the symptoms were
improved and the patient was discharged. After 1 year, the patient
was lost to follow-up. The present study was conducted with
approval from the ethics committee of Shiyan Taihe Hospital
(Shiyan, China), and with written informed consent from the
patient.
Discussion
PAM is characterized by calcifications within the
alveolar spaces, and clinical-radiological dissociation is the
hallmark of this disorder (2).
Numerous PAM patients do not present any clinical symptoms;
however, the typical presentations in symptomatic patients include
dyspnea, a nonproductive cough, sporadic hemoptysis, chest pain and
asthenia (8,14). Due to the chronic hypoxia, finger
clubbing is observed in a small portion of patients in the advanced
stages of the disease (1). Plain
chest radiography, HRCT, magnetic resonance imaging,
99mTc-methylene diphosphonate,
18F-fluorodeoxyglucose-positron emission tomography/CT
and pulmonary function tests have been commonly used to provide
diagnostic evidence for PAM (2).
However, the diagnosis of PAM can be established on the basis of
the typical radiological characteristics of the disease, which
includes interlobular septal thickening, ground-glass opacities,
calcified micronodules, pleural and subpleural calcification and
cysts (2). In the present case, HRCT
detected multiple bilateral diffuse ground-glass attenuation and
subpleural linear calcifications, which was consistent with a
previous study (9). In addition, a
lung biopsy (transbronchial or open) and BALF examination was
performed to confirm the diagnosis of PAM. Histologically, typical
findings within the alveolar spaces include numerous calcified
bodies, which are concentrically laminated with an onion skin-like
appearance (14–16). This appearance is distinct from
metastatic and dystrophic calcifications, which are located in the
interstitial or vascular compartments (2,17). In
the early stages of PAM, the lung involvement is limited, the
interlobular septa are intact and gas exchange and the pulmonary
function tests are normal (2,8).
However, as the disease progresses, the microliths gradually grow
in size and occupy a large number of alveolar spaces (2,8,15). In doing so, they make contact with
the walls, exerting pressure and causing damage that leads to the
replacement of the walls with fibrous tissue, which in turn results
in the deterioration of ventilatory and perfusion disorders
(2,8,15). In
addition, the pulmonary function tests reveal typical features of a
restrictive defect with reduced forced vital capacity (2,8). In the
present case, the pulmonary function tests suggested that the
patient had both restrictive and diffusion defects.
In previous studies, a family history of PAM was
reported in 31.8 to 37.2% of cases (1,8), and
often the parents of patients were cousins, which was indicative of
a genetic aetiology with a pattern of autosomal recessive
inheritance (1,8). First cousins are third-degree
relatives, sharing 1/8 of their genes, and with a 1/16 chance of
homozygosity by descent (1).
Consistent with this, the parents of the present patient were
cousins. A mutation in the SLC34A2 gene, which encodes the type IIb
sodium-dependent phosphate co-transporter, is considered to be
responsible for familial PAM (3).
This mutation leads to the reduced ability of the alveolar type II
cells to clean-up the phosphorus ions from the alveolar space,
which subsequently results in calcium phosphate chelation and
microlith formation in the alveolar air spaces (2,3).
To date, no effective treatment has been established
for the prevention of PAM progression. DE, which is a member of the
bisphosphonate family, has been considered a candidate drug for the
treatment of patients with PAM due to its inhibitory effect on the
precipitation of hydroxyapatite microcrystals (10). Although the effect of DE treatment on
PAM is controversial, a previous study by Ozcelik et al
(11) reported two cases of PAM that
were treated with DE (200 mg/day) for a duration of 9 and 11 years;
an evident improvement of the progression-free survival was
observed during the treatment. In addition, in one of the cases,
pulmonary calcifications gradually disappeared and no new
calcifications were formed during the 11-year treatment (11). Furthermore, Cakir et al
(12) reported the case of three
siblings (an 11-year-old boy and 4-year-old twin girls) who were
treated with DE (200 mg/day) for a 1-year period; two of the
siblings exhibited radiological improvements, whereas one did not.
Based on these results, DE may be considered a treatment option for
PAM, although further studies are required. Lung transplantation
remains the only possible treatment for end-stage cases of PAM,
although the long-term survival of such patients is uncertain
(18,19). An alternative treatment may include a
low-phosphate diet; in a previous study of a mouse model, a
low-phosphate diet prevented microlith formation in young mice and
reduced lung injury (20).
Furthermore, the burden of pulmonary calcium deposits in
established PAM was diminished within 4 weeks of a low-phosphate
diet (20). Despite these treatment
options, the long-term prognosis of patients with PAM is poor, with
advanced pulmonary fibrosis, respiratory failure, cor pulmonale and
mortality observed in end-stage PAM patients (2).
In conclusion, the present study reported a typical
case of PAM; the parents of the patient were cousins, the
predominant symptom was a dry cough and the physical examination
revealed finger clubbing. The HRCT scan showed the typical PAM
radiological appearance, including multiple bilateral diffuse
ground-glass attenuations and subpleural linear calcifications. The
mild clinical presentation was markedly dissociated from the severe
radiological appearances. A pulmonary function test suggested that
the patient had both restrictive and diffusion defects, a BALF
examination detected a calcified body in the BALF and a
transbronchial biopsy demonstrated the characteristic
intra-alveolar lamellar microiliths.
References
|
1
|
Castellana G, Castellana G, Gentile M,
Castellana R and Resta O: Pulmonary alveolar microlithiasis: Review
of the 1022 cases reported worldwide. Eur Respir Rev. 24:607–620.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferreira Francisco FA, Silva Pereirae JL,
Hochhegger B, Zanetti G and Marchiori E: Pulmonary alveolar
microlithiasis. State-of-the-art review. Respir Med. 107:1–9. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Corut A, Senyigit A, Ugur SA, Altin S,
Ozcelik U, Calisir H, Yildirim Z, Gocmen A and Tolun A: Mutations
in SLC34A2 cause pulmonary alveolar microlithiasis and are possibly
associated with testicular microlithiasis. Am J Hum Genet.
79:650–656. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tachibana T, Hagiwara K and Johkoh T:
Pulmonary alveolar microlithiasis: Review and management. Curr Opin
Pulm Med. 15:486–490. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Caffrey PR and Altman RS: Pulmonary
Alveolar Microlitbiasis Occurring in Premature Twins. J Pediatr.
66:758–763. 1965. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Krishnakurup J and Abdelsayed G: The
Calcareous Lung. Mayo Clin Proc. 86:852011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kashyap S and Mohapatra PR: Pulmonary
alveolar microlithiasis. Lung India. 30:143–147. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mariotta S, Ricci A, Papale M, De Clementi
F, Sposato B, Guidi L and Mannino F: Pulmonary alveolar
microlithiasis: Report on 576 cases published in the literature.
Sarcoidosis Vasc Diffuse Lung Dis. 21:173–181. 2004.PubMed/NCBI
|
|
9
|
Francisco FA, Rodrigues RS, Barreto MM,
Escuissato DL, Neto CAA, Silva Pereirae JL, Silva CS, Hochhegger B,
Souza AS Jr, Zanetti G and Marchiori E: Can chest high-resolution
computed tomography findings diagnose pulmonary alveolar
microlithiasis? Radiol Bras. 48:205–210. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Göcmen A, Toppare MF, Kiper N and
Büyükpamukcu N: Treatment of pulmonary alveolar microlithiasis with
a diphosphonate - preliminary results of a case. Respiration.
59:250–252. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ozcelik U, Yalcin E, Ariyurek M, Ersoz DD,
Cinel G, Gulhan B and Kiper N: Long-term results of disodium
etidronate treatment in pulmonary alveolar microlithiasis. Pediatr
Pulmonol. 45:514–517. 2010.PubMed/NCBI
|
|
12
|
Cakir E, Gedik AH, Özdemir A,
Buyukpinarbasili N, Bilgin M and Ozgen IT: Response to Disodium
Etidronate Treatment in Three Siblings with Pulmonary Alveolar
Microlithiasis. Respiration. 89:583–586. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Enright PL and Sherrill DL: Reference
equations for the six-minute walk in healthy adults. Am J Respir
Crit Care Med. 158:1384–1387. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lauta VM: Pulmonary alveolar
microlithiasis: An overview of clinical and pathological features
together with possible therapies. Respir Med. 97:1081–1085. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Barnard NJ, Crocker PR, Blainey AD, Davies
RJ, Ell SR and Levison DA: Pulmonary alveolar microlithiasis. A new
analytical approach. Histopathology. 11:639–645. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jönsson ÅL, Simonsen U, Hilberg O and
Bendstrup E: Pulmonary alveolar microlithiasis: Two case reports
and review of the literature. Eur Respir Rev. 21:249–256. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chan ED, Morales DV, Welsh CH, McDermott
MT and Schwarz MI: Calcium deposition with or without bone
formation in the lung. Am J Respir Crit Care Med. 165:1654–1669.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Samano MN, Waisberg DR, Canzian M, Campos
SV, Pêgo-Fernandes PM and Jatene FB: Lung transplantation for
pulmonary alveolar microlithiasis: A case report. Clinics (Sao
Paulo). 65:233–236. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Borrelli R, Fossi A, Volterrani L and
Voltolini L: Right single-lung transplantation for pulmonary
alveolar microlithiasis. Eur J Cardiothorac Surg. 45:e402014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Saito A, Nikolaidis NM, Amlal H, Uehara Y,
Gardner JC, LaSance K, Pitstick LB, Bridges JP, Wikenheiser-Brokamp
KA, McGraw DW, et al: Modeling pulmonary alveolar microlithiasis by
epithelial deletion of the Npt2b sodium phosphate cotransporter
reveals putative biomarkers and strategies for treatment. Sci
Transl Med. 7:313ra1812015. View Article : Google Scholar : PubMed/NCBI
|