Introduction
Acute lung injury (ALI) is characterized by damage
to the alveolar epithelial and capillary endothelial cells
(1). ALI can be induced by various
factors, such as pulmonary alveolar hemorrhage, shock and
transfusion, resulting in diffuse pulmonary interstitial and
alveolar edema, refractory hypoxemia and respiratory distress
(2–4). Although the pathogenesis of ALI remains
unclear, Toll-like receptor (TLR)-mediated systemic inflammatory
response may be involved (5–8).
TLRs are a class of cellular receptors that
recognize pathogen-associated molecular patterns in innate
immunity. TLRs associate the innate and adaptive immunity (9). The activation of TLR2 and TLR4
signaling cascades leads to the production of cytokines and
co-stimulatory factors (9,10). The release of inflammatory cytokines
by various effector cells is an important factor contributing to
ALI (11). Ligands bind TLR4, which
in turn activates nuclear factor (NF)-κB through a myeloid
differentiation protein 88 (MyD88)-dependent signaling pathway.
This ultimately stimulates the expression of pro-inflammatory
cytokines, such as interleukin (IL)-1, IL-6 and tumor necrosis
factor (TNF)-α, which leads to pathological changes in ALI
(12).
Anticholinergic drugs are an effective and widely
accepted treatment for ALI (13,14).
They decrease pro-inflammatory effects by resisting M cholinergic
function, inhibiting choline and preventing the release of
5-hydroxytryptamine from epithelial cells and of neutrophilic
chemotactic factor from lung alveolus macrophages (15). Anisodamine treatment of
lipopolysaccharide (LPS)-induced ALI in rats has been found to
inhibit the expression of pro-inflammatory cytokines, including
TNF-α and IL-8 (16). Penehyclidine
(PHC) is a novel anticholinergic agent that selectively acts on
receptors of M1, M3, ganglion and central N1 cells to decrease the
TNF-α expression levels and NF-κB activity, inhibiting the
inflammatory response (17).
Furthermore, PHC treatment decreased TNF-α and IL-6 expression
levels and suppressed the inflammatory response following
extracorporeal circulation (18).
However, the mechanism underlying the effects of PHC treatment on
the inflammatory response remains unclear. To the best of our
knowledge, few studies have investigated the effect of PHC on
endotoxin-induced ALI (19,20). Therefore, the aim of the present
study was to investigate whether PHC treatment was able to
alleviate ALI in rats. The expression levels of TLR2/4 and NF-κB
activation were also examined in the lung tissue.
Materials and methods
Animals and reagents
A total of 60 Sprague-Dawley rats (age, 8–10 weeks;
weight, 200–220 g) were used in the present study (Identification
Institute of Pharmaceutical and Biological Products of Beijing,
Beijing, China). Rats were maintained in the animal laboratory at
the Tuberculosis and Thoracic Tumor Research Institute (Beijing,
China) at room temperature (16–26°C) and relative humidity
(60%)with a 12:12 h light/dark cycle. A total of 12 100% fresh air
changes were made per hour. Rats had ad libitum access to a
standard diet and water. All experimental procedures in the present
study adhered to the Principles of Experimental Animal Care and Use
in accordance with the guidelines of the Helsinki Declaration and
was approved by the Ethics Committee of the Beijing Chest Hospital,
Capital Medical University. Ethical approval was obtained from the
Institutional Review Board and Ethics Committee of the Tuberculosis
and Thoracic Tumor Research Institute. LPS was purchased from
Sigma-Aldrich (St. Louis, MO, USA; cat. no. L2880). PHC injection
solution was purchased from Chengdu Lisite Pharmaceutical, Co.,
Ltd. (Chengdu, China; batch, H20020606). Rat TNF-α and IL-6 ELISA
kits were purchased from Sigma-Aldrich.
Animal models and grouping
Rats were randomly divided into five equal groups
(n=12) and intraperitoneally injected with 2 ml 0.9% saline
(control group), 2 ml 0.9% saline containing LPS (8 mg/kg body
weight; LPS-alone group), or three groups injected with 2 ml 0.9%
saline containing LPS (8 mg/kg body weight) and different PHC
concentrations (0.3, 1, or 3 mg/kg body weight). PHC was
administered 1 min following the LPS injection. At 6 h after drug
administration, the rats were sacrificed via cervical dislocation
following anesthetization via intraperitoneal injection of sodium
pentobarbital (35 mg/kg body weight; Tianjin Jinyao Amino Acid Co.,
Ltd., Beijing, China). Whole blood (1 ml) was harvested from the
ventricles and placed into disposable vacuum vessels to measure the
serum levels of TNF-α and IL-6. Left lung tissue samples were
obtained, homogenized, snap-frozen and stored in liquid nitrogen
until further use. The remaining lung tissue samples were fixed in
10% formaldehyde solution.
ELISA
The levels of TNF-α and IL-6 in the blood samples
were detected using ELISA kits according to the manufacturer's
protocol. Briefly, 100 µl serum was added to a 96-well plate
pre-coated with TNF-α or IL-6 antibody and incubated at 4°C
overnight. Following washing three times with washing buffer, 100
µl 1x biotinylated detection antibody was added to each well and
incubated at room temperature for 1 h with gentle shaking,
following with incubation with 100 µl horseradish peroxidase (HRP)
conjugated-streptavidin solution for 45 min at room temperature.
Subsequently, the plate was treated with 100 µl
3,3′,5,5′-tetramethylbenzidine substrate, incubated for 30 min at
room temperature and then the reaction was terminated with 50 µl
Stop solution per well. Immediately, the plate was read at 450 nm
using a microplate reader (Molecular Devices, LLC, Sunnyvale, CA,
USA).
Western blot analysis of NF-κB p65
expression
Nuclear extracts of the left lung tissue samples
(0.2 g) were extracted using buffer A [10 mM HEPES pH 7.9, 10 mM
KCl, 1.5 mM MgCl2, 1 mM dithiothreitol (DTT), 0.4 mM
phenylmethanesulfonyl fluoride (PMSF)] and buffer B (20 mM HEPES pH
7.9, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM DTT, 0.4 mM PMSF,
10 µg/ml leupeptin, 1 µg/ml aprotinin, 25% glycerol). Nucleoprotein
concentrations were determined using a NanoDrop 2000
spectrophotometer (Thermo Fisher Scientific, Inc., Waltham, MA,
USA) and adjusted to 0.2 µg/μl. Equal amounts (20 µg) of protein
samples were resolved by 4–12% SDS-PAGE, and transferred to
nitrocellulose membranes (Beijing Dingguo Biological Technology
Co., Ltd., Beijing, China). Following blocking with 5%
milk/Tris-buffered saline with Tween 20 (TBST; Sigma-Aldrich) at
room temperature for 1 h, the membranes were incubated at 4°C
overnight with rat anti-NF-κB p65 polyclonal antibody (1:300;
BA0610) in 5% milk/TBST, then incubated with HRP-conjugated goat
anti-rabbit immunoglobulin G (1:4,000; BA1054; both Wuhan Boster
Biological Technology, Ltd., Wuhan, China) in 5% milk/TBST. Blots
were analyzed using a gel imager (Gel Doc XR; Bio-Rad Laboratories,
Inc., Hercules, CA, USA). Band Leader 3.0 software (Magnitec, Ltd.,
Tel Aviv, Israel) was used for semi-quantitative analysis of
protein expression.
Reverse transcription-quantitative
polymerase chain reaction (PCR) analysis of TLR2/4 expression in
the lung tissue samples
A total of 100 mg left lung tissue sample was lysed
with TRIzol® (Invitrogen; Thermo Fisher Scientific,
Inc.,) and RNA was extracted according to the manufacturer's
protocol. An equal amount of total RNA (1 µg) was used for each
sample to synthesize cDNA, prior to PCR analysis. The following
primers were designed using Primer 3 software and synthesized by
SBS Genetech Co., Ltd., (Shanghai, China): TLR4, forward
5′-GGCATGGCATGGCTTAAACC-3′ and reverse
5′-AATTCTCCCAAGATCAACCGATG-3′; TLR2, forward
5′-GGGCTGACCTCTCTCAACGA-3′ and reverse
5′-CAGTTCCAAGATGTAACGCAACA-3′; and β-actin, forward
5′-ACTGCCGCATCCTCTTCCTC-3′ and reverse 5′-AAGCATTTGCGGTGCACGA-3′.
Thermal cycling was performed using SYBR Green PCR Master Mix on a
MyCycler (both Bio-Rad Laboratories, Inc.) as follows: TLR4, 95°C
for 2 min, followed by 30 cycles of 95°C for 1 min, 59°C for 1 min
and 72°C for 45 sec, with a final hold at 72°C for 10 min; TLR2,
95°C for 2 min, followed by 30 cycles of 95°C for 1 min, 60°C for 1
min and 72°C for 45 sec, with a final hold at 72°C for 10 min; and
β-actin: 95°C for 2 min, followed by 28 cycles of 95°C for 1 min,
59°C for 1 min and 72°C for 45 sec, with a final hold at 72°C for 7
min. Amplification products were resolved by 2% agarose gel
electrophoresis, and imaged using a Gel Doc EZ imaging system, and
analyzed using GIS gel analysis software (Gel Doc XR; Bio-Rad
Laboratories, Inc.). TLR2 and TLR4 mRNA expression levels were
normalized to those of β-actin.
Examination of lung tissue
pathology
Left lung tissue samples were obtained and embedded
in paraffin wax, sectioned (0.5×0.5 cm), and then stained with
hematoxylin and eosin (Beijing Dingguo Biological Technology Co.,
Ltd.). The tissue was analyzed using a light microscope (DM4; Leica
Microsystems GmbH, Wetzlar, Germany) at ×400 magnification.
Smith-scoring (21) was performed on
the tissue samples. Lung pathology was assessed for edema and
alveolar and interstitial inflammation and hemorrhage scored on a
0- to 4-point scale: 0, no injury; 1, injury in 25% of the field;
2, injury in 50% of the field; 3, injury in 75% of the field; and
4, injury throughout the field (>75%) (21).
Statistical analysis
All values are expressed as the mean ± standard
deviation. One-way analysis of variance was performed using SPSS
13.0 (SPSS Inc., Chicago, IL, USA). P<0.05 was considered to
indicate a statistically significant result.
Results
LPS-induced endotoxemia in rats
LPS-induced endotoxemia rat models are recognized as
valid experimental models for clinical and experimental
applications (22). In the ALI model
established in the present study, rats injected with LPS exhibited
mental fatigue, curling-up behavior, a delayed response, ocular
discharge, and steady breathing compared with the control rats.
Pathological analysis demonstrated that the lung tissue samples in
the LPS group contained lesions, vascular engorgements and alveolar
epithelial cells with edema (Fig.
1). Lymphocyte and neutrophil infiltration was observed in the
alveolar wall, peribronchiolar and interstitial lungs, as well as
serous effusion and extravasated red cells in the alveolar
cavities. These data indicate that a rat model of endotoxemia was
successfully established.
Smith-scoring and examination of lung
tissue pathology
Tissue analysis was performed using a light
microscope (magnification, ×400). The pulmonary morphology was
normal in the control group (Fig.
1A). By contrast, pulmonary consolidations, vascular
engorgements, alveolar epithelial cells with edema, lymphocyte and
neutrophil infiltration in the alveolar wall, peribronchiolar and
interstitial lungs, serous effusion and extravasated red cells in
the alveolar cavities were observed in the LPS-treated group
(Fig. 1B). Lung tissue inflammatory
changes, inflammatory cell infiltration, widening of the alveolar
septum and hyperemia were observed in the rats treated with high
doses of PHC (1 and 3 mg/kg body weight); however, there was no
serous effusion or extravasated red cells in the alveolar space
(Fig. 1C–E). Low dose PHC (0.3 mg/kg
body weight) had no significant effect. The Smith-scores in all of
the LPS-induced groups were increased compared with the control
group (P<0.05; Fig. 1F), which
indicated edema of lung epithelial cells and infiltration of
lymphocytes and neutrophils into the lung parenchyma (1,21).
Scores in the PHC-treated groups (1 and 3 mg/kg body weight) were
decreased compared with the LPS group (P<0.05).
Effects of PHC treatment on TNF-α and
IL-6 expression levels in the endotoxemia rat model
LPS markedly increased the expression levels of
TNF-α, which is primarily produced by monocytes and macrophages.
Following the intraperitoneal injection of LPS, TNF-α is the first
cytokine to increase to detectable levels (11). IL-6, which is primarily generated by
T cells, endothelial cells and monocytes, mediates acute responses
contributing to lung injury during systemic inflammatory response
syndrome (SIRS). Trauma, shock, infection and surgical procedures
have been shown to increase IL-6 expression levels (18). Increased expression levels of IL-6
activate complement activity and C-reactive protein, causing
cellular damage, adhesion factor production, and activation of
glial cells, vascular endothelial cells and lymphocytes, leading to
an aggravated inflammatory response (23). In the present study, serum levels of
TNF-α and IL-6 significantly increased following intraperitoneal
injection of LPS compared with the levels in the control rats. PHC
treatment at doses of 1 and 3 mg/kg body weight following LPS
injection resulted in decreased serum levels of TNF-α and IL-6
compared with the group treated with LPS alone (P<0.05; Fig. 2); whereas 0.3 mg/kg PHC had no
significant effect.
TLR2/4 mRNA expression levels in the
lung tissue of LPS-induced rats
TLR2 and TLR4 mRNA expression levels in the lung
tissue samples were increased in the LPS-induced rats compared with
the levels in the control group (P<0.05; Fig. 3). PHC treatment significantly
decreased TLR2 and TLR4 mRNA expression levels in lung tissue
samples at a dose of 1 and 3 mg PHC/kg body weight, although no
statistically significant difference was observed between the rats
treated with 0.3 mg PHC/kg body weight and those treated with LPS
alone (Fig. 3).
 | Figure 3.(A) TLR2 and TLR4 mRNA expression in
the left lung tissue samples of LPS-treated rats, as determined
using reverse transcription-quantitative polymerase chain reaction
and agarose gel electrophoresis. β-actin was used as the loading
control. (B) TLR2 and (C) TLR4 mRNA expression levels, quantified
from the gels based on the average absorbance values of the bands.
TLR mRNA expression levels are presented as the mean ± standard
deviation of TLR/β-actin for each sample. Groups: Lane 1, marker;
lane 2, control (saline-treated); lane 3, LPS only; lane 4, 0.3 mg
PHC/kg body weight + LPS; lane 5, 1 mg PHC/kg body weight + LPS;
lane 6, 3 mg PHC/kg body weight + LPS. *P<0.05 vs. the saline
control; #P<0.05 vs. the LPS-treated groups. TLR2/4,
toll-like receptor 2/4; LPS, lipopolysaccharide; PHC,
penehyclidine. |
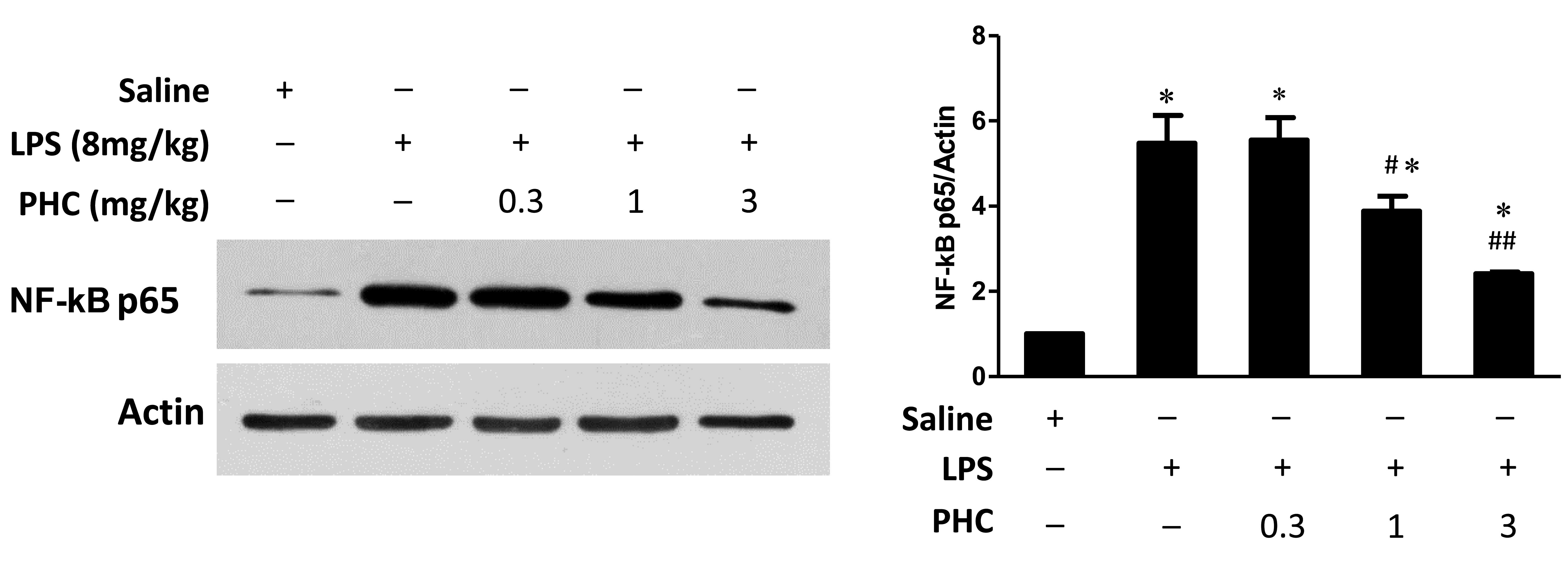
Effects of PHC treatment on nuclear
NF-κB p65 expression levels in endotoxemic rats
Cellular inflammatory signaling pathways may be
regulated by activated nuclear factors. In particular, NF-κB is an
important nuclear transcription factor during this process. NF-κB
mediates various inflammatory cellular events, including immune
cell activation, T and B lymphocyte development, stress response
and apoptosis (24). In the current
study, NF-κB p65 protein expression levels were increased in the
lung tissue samples in the LPS group, compared with those in the
control group (Fig. 4). However, PHC
treatment resulted in a dose-dependent decrease in NF-κB p65
expression levels in lung tissue samples, as compared with the LPS
group (1 and 3 mg/kg groups, P<0.05).
Discussion
Cytokine release in SIRS is involved in the
pathogenesis of ALI (25). The
response to intravenous or intraperitoneal injection of LPS is
similar to the response to endotoxin release in severe
gram-negative bacterial infection, which triggers the inflammatory
response (26). In the present
study, intraperitoneal injection of LPS was used to establish a rat
model of endotoxemia.
TLRs mediate the innate immunity and play important
roles in inflammation, signal transduction, apoptosis and
tumorigenesis (27). As a specific
receptor of LPS, TLR4 transmits the inflammatory signal induced by
LPS (28). The LPS-binding protein
(LBP) is a plasma protein that is synthesized and secreted by the
liver. When LBP binds lipid A in LPS, a CD14-LPS complex forms. LBP
is involved in phospholipid transfer, and disassociates LPS from
its oligomeric state into monomers (29). Depolymerized LPS binds to TLR4,
leading to polymerization and activation of TLR4.
MD-2 is a secretory protein lacking a transmembrane
region that assists in the binding of the cytoplasmic tail of TRL4
to the TLR4 receptor-binding domain of the MyD88 adaptor protein
(30). MyD88 binds IL-1
receptor-associated kinase (IRAK) via its death domain. IRAK is
autophosphorylated, and TNF-α receptor-associated factor-6 (TRAF-6)
is activated. Subsequent activation of the mitogen-binding protein
kinase family leads to the activation of NF-κB-inducing kinase
(NIK) (31). IκB kinase is activated
and phosphorylated by NIK, leading to its degradation and NF-κB
release. NF-κB then translocates to the nucleus to initiate
transcription (32). TRAF-6 also
activates mitogen-activated protein kinase kinase kinase (MAPKKK),
MAPKK, extracellular signal-regulated kinase, p38 and c-Jun
N-terminal kinase/stress-activated protein kinase signaling,
leading to the activation of transcription factor-activating
protein-1 (AP-1) (33). Activation
of NF-κB and AP-1 leads to increased expression levels of
inflammatory factors, such as TNF-α, IL-1, IL-6 and nitric oxide,
resulting in ALI/acute respiratory distress syndrome.
TLR2 predominantly mediates the activation of cells
by gram-positive bacteria. LPS and cytokines induced by LPS
upregulate TLR2 mRNA expression in macrophages (34). Therefore, although TLR2 does not bind
LPS directly, it may play an important role in enhancing
stress-induced lung injury by functioning as a secondary receptor.
TLR2 expression is known to be regulated by NF-κB (35). In addition, LPS-induced activation of
TLR4 leads to NF-κB activation, and has been shown to induce the
synthesis of large quantities of inflammatory factors and to
upregulate TLR2 mRNA expression, increasing the inflammatory
response (31,36,37). The
results demonstrated that PHC treatment may alleviate ALI caused by
hemorrhagic shock by decreasing TLR4 mRNA expression levels in the
lung, inhibiting NF-κB activity and reducing TNF-α expression.
In the present study, the results suggested that PHC
treatment of LPS-induced rats resulted in decreased TLR4 and TLR2
mRNA expression levels and NF-κB activity levels in the lung tissue
samples, as well as reduced serum levels of TNF-α and IL-6. The
extent of lung tissue damage was reduced, consistent with the
molecular results, suggesting that PHC may inhibit rat inflammatory
responses and alleviate ALI by acting on the inflammatory response
signaling pathways in a dose-dependent manner.
There were several limitations to the present study.
Firstly, a limited number of animals were used, and therefore
additional studies should be performed to further examine the
results. Secondly, clinical data demonstrating the results
presented here would be beneficial. Thirdly, additional experiments
such as investigations of the exact mechanism underlying TLR ligand
recognition and of the transcriptional regulation of TLR4 and TLR
activation signaling pathway that links the innate and acquired
immunity should be performed to provide further evidence for the
results presented in the current study.
In conclusion, the present study demonstrated that
treatment with 1 and 3 mg/kg PHC significantly attenuated
LPS-induced upregulation of lung TLR2 and TLR4 mRNA and decreased
NF-κB activation and serum TNF-α and IL-6 levels, resulting in the
alleviation of inflammation. Therefore, PHC may have a protective
role in LPS-induced ALI in rats and may be a novel candidate for
the treatment of inflammation-related lung injury.
Acknowledgements
The authors of the present study would like to thank
Dr Ting-Ming Cao and Dr Yang Liu (both Capital Medical University)
for their technical assistance.
References
|
1
|
Luh SP and Chiang CH: Acute lung
injury/acute respiratory distress syndrome (ALI/ARDS): The
mechanism, present strategies and future perspectives of therapies.
J Zhejiang Uni Sci B. 8:60–69. 2007. View Article : Google Scholar
|
|
2
|
Chen H, Zhang L, Jin Z, Jin E, Fujiwara M,
Ghazizadeh M, Asoh S, Ohta S and Kawanami O: Anti-apoptotic PTD-FNK
protein suppresses lipopolysaccharide-induced acute lung injury in
rats. Exp Mol Pathol. 83:377–384. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu YY, Li LF, Yang CT, Lu KH, Huang CC,
Kao KC and Chiou SH: Suppressing NF-κB and NKRF pathways by induced
pluripotent stem cell therapy in mice with ventilator-induced lung
injury. PLoS One. 8:e667602013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lucas R, Verin AD, Black SM and Catravas
JD: Regulators of endothelial and epithelial barrier integrity and
function in acute lung injury. Biochem Pharmacol. 77:1763–1772.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Andonegui G, Bonder CS, Green F, Mullaly
SC, Zbytnuik L, Raharjo E and Kubes P: Endothelium-derived
Toll-like receptor-4 is the key molecule in LPS-induced neutrophil
sequestration into lungs. J Clin Invest. 111:1011–1020. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Andonegui G, Goyert SM and Kubes P:
Lipopolysaccharide-induced leukocyte-endothelial cell interactions:
A role for CD14 versus toll-like receptor 4 within microvessels. J
Immunol. 169:2111–2119. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ayala A, Chung CS, Lomas JL, Song GY,
Doughty LA, Gregory SH, Cioffi WG, LeBlanc BW, Reichner J, Simms HH
and Grutkoski PS: Shock-induced neutrophil mediated priming for
acute lung injury in mice: Divergent effects of TLR-4 and
TLR-4/FasL deficiency. Am J Pathol. 161:2283–2294. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Basu S and Fenton MJ: Toll-like receptors:
Function and roles in lung disease. Am J Physiol Lung Cell Mol
Physiol. 286:L887–L892. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Takeda K, Kaisho T and Akira S: Toll-like
receptors. Annu Rev Immunol. 21:335–376. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hirschfeld M, Weis JJ, Toshchakov V,
Salkowski CA, Cody MJ, Ward DC, Qureshi N, Michalek SM and Vogel
SN: Signaling by toll-like receptor 2 and 4 agonists results in
differential gene expression in murine macrophages. Infect Immun.
69:1477–1482. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Takaoka Y, Goto S, Nakano T, Tseng HP,
Yang SM, Kawamoto S, Ono K and Chen CL: Glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) prevents lipopolysaccharide (LPS)-induced,
sepsis-related severe acute lung injury in mice. Sci Rep.
4:52042014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Perros F, Lambrecht BN and Hammad H: TLR4
signalling in pulmonary stromal cells is critical for inflammation
and immunity in the airways. Respir Res. 12:1252011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xiu RJ, Hammerschmidt DE, Coppo PA and
Jacob HS: Anisodamine inhibits thromboxane synthesis, granulocyte
aggregation and platelet aggregation. A possible mechanism for its
efficacy in bacteremic shock. JAMA. 247:1458–1460. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang Y, Chen Q, Liu S, Huang Y, Liu L, Wu
X, Chen G, Jin J, Teng G and Qiu H: Effects of recruitment
maneuvers with PEEP on lung volume distribution in canine models of
direct and indirect lung injury. Mol Biol Rep. 41:1325–1333. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Disse B: Antimuscarinic treatment for lung
diseases from research to clinical practice. Life Sci.
68:2557–2564. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang Z, Wang C, Tao J, Xiong J, Wan C and
Zhou F: Effect of early hemofiltration on pro- and
anti-inflammatory responses and multiple organ failure in severe
acute pancreatitis. J Huazhong Univ Sci Technol Med Sci.
24:456–459. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Feng S, Ge Y, Yang J, Duan M and Xu J:
Effects of penehyclidine hydrochloride on lung inflammatory
response in septic rats. Zhong Hua Ma Zui Xue Za Zhi Bian Ji Bu.
28:63–67. 2008.(In Chinese).
|
|
18
|
Huang C, He J, Chen Y, Zhang Y and Chen C:
Penehyclidine hydrochloride inhibits the LPS-induced inflammatory
response in microglia. J Surg Res. 188:260–267. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lei L, Wang Y, Jia B, Zhan J and Wang C:
Protective effect of penehyclidine hydrochloride on lung injury in
mice with sepsis and its mechanism. Zhongguo Wei Zhong Bing Ji Jiu
Yi Xue. 19:623–625. 2007.(In Chinese). PubMed/NCBI
|
|
20
|
Li BQ, Sun HC, Nie SN, Shao DB, Liu HM and
Qian XM: Effect of penehyclidine hydrochloride on patients with
acute lung injury and its mechanisms. Chin J Traumatol. 13:329–335.
2010.PubMed/NCBI
|
|
21
|
Smith KM, Mrozek JD, Simonton SC, Bing DR,
Meyers PA, Connett JE and Mammel MC: Prolonged partial liquid
ventilation using conventional high-frequency ventilatory
techniques: Gas exchange and lung pathology in animal model of
respiratory distress syndrome. Crit Care Med. 25:1888–1897. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rojas M, Woods CR, Mora AL, Xu J and
Brigham KL: Endotoxin-induced lung injury in mice:structural,
functional, and biochemical responses. Am J Physiol Lung Cell Mol
Physiol. 288:L333–L341. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Petersen AM and Pedersen BK: The
anti-inflammatory effect of exercise. J Appl Physiol. 98:1154–1162.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Oeckinghaus A and Ghosh S: The NF-κB
family of transcription factors and its regulation. Cold Spring
Harb Perspect Biol. 1:a0000342009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Aikawa N, Ishizaka A, Hirasawa H,
Shimazaki S, Yamamoto Y, Sugimoto H, Shinozaki M, Taenaka N, Endo
S, Ikeda T and Kawasaki Y: Reevaluation of the efficacy and safety
of the neutrophil elastase inhibitor, Sivelestat, for the treatment
of acute lung injury associated with systemic inflammatory response
syndroma phase IV study. Pulm Pharmacol Ther. 24:549–554. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Haddad JJ: Nuclear factor (NF)-kappa B
blockade attenuates but does not abrogate LPS-mediated interleukin
(IL)-1 beta biosynthesis in alveolar epithelial cells. Biochem
Biophys Res Commun. 293:252–257. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ben-Addi A, Mambole-Dema A, Brender C,
Martin SR, Janzen J, Kjaer S, Smerdon SJ and Ley SC: IκB
kinase-induced interaction of TPL-2 kinase with 14-3-3 is essential
for Toll-like receptor activation of ERK-1 and −2 MAP kinases. Proc
Natl Acad Sci USA. 111:E2394–E2403. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chow JC, Young DW, Golenbock DT, Christ WJ
and Gusovsky F: Toll-like receptor-4 mediates
lipopolysaccharide-induced signal transduction. J Biol Chem.
274:10689–10692. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Erridge C, Kennedy S, Spickett CM and Webb
DJ: Oxidized phospholipid inhibition of toll-like receptor (TLR)
signaling is restricted to TLR2 and TLR4: Roles for CD14,
LPS-binding protein and MD2 as targets for specificity of
inhibition. J Biol Chem. 283:24748–24759. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shimazu RA, Kashi S, Ogata H, Nagai Y,
Fukudome K, Miyake K and Kimoto M: MD-2, a molecule that confer
1ipopolysaccharide responsiveness on Toll-like receptor 4. J Exp
Med. 189:1777–1782. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Akira S: Toll-like receptor signaling. J
Biol Chem. 278:38105–38108. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Brikos C, Wait R, Begum S, O'Neill LA and
Saklatvala J: Mass spectrometric analysis of the endogenous type I
interleukin-1 (IL-1) receptor signaling complex formed after IL-1
binding identifies IL-1RAcP, MyD88 and IRAK-4 as the stable
components. Mol Cell Proteomics. 6:1551–1559. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kopp EB and Medzhitov R: The Toll-receptor
family and control innate immunity. Curr Opin Immunol. 11:13–18.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu Y, Wang Y, Yamakuchi M, Isowaki S,
Nagata E, Kanmura Y, Kitajima I and Maruyama I: Upregulation of
toll-like receptor 2 gene expression in macrophage response to
peptidoglycan and high concentration of lipopolysaccharide is
involved in NF-κB activation. Infect Immun. 69:2788–2796. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Musikacharoen T, Matsuguchi T, Kikuchi T
and Yoshikai Y: NF-kappa B and STAT5 play important roles in the
regulation of mouse Toll-like receptor 2 gene expression. J
Immunol. 166:4516–4524. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chow JC, Young DW, Golenbock DT, Christ WJ
and Gusovsky F: Toll-like receptor-4 mediates
lipopolysaccharide-induced signal transduction. J Biol Chem.
274:10689–10692. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gilmore TD: Introduction to NF-kappaB:
Players, pathways, perspectives. Oncogene. 25:6680–6684. 2006.
View Article : Google Scholar : PubMed/NCBI
|