Introduction
Viral myocarditis is a potentially lethal infection
often resulting in arrhythmia, heart failure, dilated
cardiomyopathy and mortality (1).
There are three phases of viral myocarditis. During the early
phase, viral infection and replication cause direct damage to the
myocardial cells. Autoimmune myocarditis, which is characterized by
autoimmune injury induced by the viral infection, is the second
stage and involves the expression and secretion of numerous
inflammatory cytokines and chemokines. Finally, in the last phase,
inflammation progressively subsides, although the viral genomes
persist in the heart (2). Myocardial
fibrosis and pathological remodeling, which lead to impaired heart
function, may occur during the early and late stages of viral
myocarditis. Coxsackievirus of group B3 (CVB3) is one of the most
common cardiotropic viruses and is among numerous known etiologies
for myocarditis (3,4). In the pathology of CVB3-induced
myocarditis, cardiac fibroblasts are an important contributor to
virus replication-mediated aggravation of myocarditis (5); they are central mediators of
inflammatory and fibrotic myocardial remodeling in the injured and
failing heart (6).
Adenosine monophosphate-activated protein kinase
(AMPK) is a key regulator of cellular energy homeostasis, and is
involved in multiple anabolic and catabolic signaling pathways in
myocardial cells (7). It has been
reported that the long-term activation of AMPK was able to
attenuate angiotensin II-induced or pressure-overload cardiac
hypertrophy (8,9). In addition, the association between
AMPK and virus infection has been investigated, and it was
demonstrated that activation of AMPK was able to restrict CVB3
replication by inhibiting lipid accumulation (10). However, it remains unclear whether
AMPK has an effect on CVB3-induced inflammatory cardiomyopathy and
myocardial fibrosis. Therefore, the present study aimed to
investigate the effect of AMPK activation on cardiac fibroblast
proliferation following CVB3 infection.
Materials and methods
Reagents
5-Aminoimidazole-4-carboxamide-ribonucleoside
(AICAR) was purchased from Toronto Research Chemicals (Toronto, ON,
Canada). Compound C was purchased from EMD Millipore (San Diego,
CA, USA). Antibodies against phospho-AMPK α-Thr172
(p-AMPKα-Thr172) were from Cell Signaling Technology,
Inc. (Beverly, MA, USA). Antibodies against glyceraldehyde
3-phosphate dehydrogenase (GAPDH) were from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA).
Determination of virus titers
The CVB3 Nancy strain (cat. no. VR30; ATCC,
Manassas, VA, USA) was propagated four times in HeLa cells (cat.
no. CCL-2; ATCC). The infected HeLa cells were broken up by three
cycles of freezing and thawing. Then, debris was removed, and
serial dilutions (10-fold) of the supernatants were prepared in
infection medium, which comprised minimal essential medium (GE
Healthcare Life Sciences, Logan, UT, USA) for HeLa cells or
fibroblasts, 2% fetal bovine serum (FBS; GE Healthcare Life
Sciences), 30 mM MgCl2, 100 U/ml penicillin, 100 µg/ml streptomycin
sulfate (both Gen-View Scientific, Inc., El Monte, CA, USA) and 2
mM glutamine (Amresco LLC, Solon, OH, USA). Subsequently, the
samples were transferred onto subconfluent monolayers of HeLa cells
grown in 96-well culture plates containing 100 ml infection medium.
Following incubation at 37°C for 24 h, cells were stained with 0.5%
crystal violet (in water) for 5 min. The 50% tissue culture
infectious dose (TCID50) was calculated. The tissue culture
infectious dose infecting 50% of the cells was calculated using the
Reed-Muench method (11).
Cell isolation and culture
Cardiac fibroblasts were isolated from neonatal
Sprague Dawley rats (age, 1 day). Minced ventricles were digested
with 0.1% trypsin and 0.03% collagenase II, and the cells were
collected and placed in 10-cm cell culture plates containing 10 ml
Dulbecco's modified Eagle's medium (DMEM; GE Healthcare Life
Sciences) supplemented with 1% penicillin-streptomycin and 10% FBS.
After 60 min in a 37°C incubator, cardiac fibroblasts attached to
the culture plates. Then, the fibroblasts were washed twice and
cultured in DMEM with 10% FBS at 37°C for 48 h until they reached
confluence. Subsequently, cultured cells were confirmed to be pure
cardiac fibroblasts by morphological inspection. All the cardiac
fibroblasts were incubated in DMEM with 2% FBS for 24 h before the
experiments were performed. The experiments were as follows: i) To
explore the effects of CVB3 on cardiac fibroblasts, neonatal
cardiac fibroblasts (5×104 cells/well) in serum-free
DMEM medium were infected with CVB3 (100 TCID50) for 24, 48 and 72
h; ii) To explore the effect of activated AMPK on cardiac
fibroblast proliferation and collagen secretion, the fibroblasts
were pretreated with 1 mmol/l AICAR (a specific AMPK activator) for
2 h, then infected with CVB3 (100 TCID50) for 48 h; and iii) the
fibroblasts were preincubated with 1 µmol/l Compound C (a specific
AMPK inhibitor) for 30 min, then treated with AICAR (1 mmol/l) and
CVB3 (100 TICD50) in succession. The animal care and experimental
protocols were in compliance with the Animal Management Rule of the
People's Republic of China (Ministry of Health, China; document no.
552; http://www.gov.cn/gongbao/content/2011/content_1860757.htm)
and the study was approved by the Animal Care Committee of the
Third Affiliated Hospital of Nantong University (Wuxi, China).
Cell proliferation assay
Cell counting and detection of
5-bromo-2′-deoxyuridine (BrdU) incorporation were used to assess
cellular proliferation. Briefly, cells were detached using 0.25%
trypsin (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). Following centrifugation at 3,000 × g for 3 min at 4°C, the
cells were resuspended in phosphate-buffered saline (PBS), after
which 25 µl aliquots of the cell suspension were mixed with an
equal volume of 0.4% trypan blue dye (Sigma-Aldrich, St. Louis, MO,
USA) at room temperature for 3 min. Finally, the samples were
placed in a hemocytometer (Neubauer improved cell counting chamber;
Paul Marienfeld GmbH & Co. KG; Lauda-Königshofen, Germany) and
counted under an Olympus IX73 microscope (Olympus Corporation,
Tokyo, Japan). The BrdU incorporation assay was performed using a
colorimetric BrdU cell proliferation enzyme-linked immunosorbent
assay (ELISA) kit for rats (Roche, Mannheim, Germany) according to
the manufacturer's protocol.
Flow cytometric assessment of cell
cycle
The cell samples were washed twice with cold PBS,
then fixed with 70% ethanol. After allowing to stand at 4°C
overnight, the samples were resuspended with PBS including 0.1
mg/ml RNase (Sigma-Aldrich). Subsequently, cells were incubated at
37°C for 30 min. After that, 50 mg/ml propidium iodide was used to
suspend the cells. Finally, the cell samples were analyzed by flow
cytometry (FACSCalibur™; BD Biosciences, San Jose, CA, USA), and
the cell cycle distribution, specifically the percentage of cells
in the S phase, was determined using ModFit LT cell cycle analysis
software (Verity Software House, Topsham, ME, USA).
Hydroxyproline measurement
A rat hydroxyproline ELISA kit (Kaibo Biochemical
Reagents Co., Ltd., Shanghai, China) was used to detect collagen
levels on the basis of the content of its major component,
hydroxyproline. Hydroxyproline concentration was calculated
according to the OD value at 450 nm provided using an ELISA plate
reader (Model 550; Bio-Rad Laboratories, Inc., Hercules, CA,
USA).
Western blot analysis
Cell samples were washed once with cold PBS
following the treatment. Subsequently, buffer [100 mmol/l NaCl, 10
mmol/l sodium pyrophosphate, 1 mmol/l sodium vanadate, 50 mmol/l
NaF, 5 mmol/l ethylenediamine tetra-acetic acid, 1% sodium
deoxycholate, 20 mmol/l Tris-HCl (pH 7.4), 0.1% sodium dodecyl
sulfate (SDS), 1 mmol/l phenylmethylsulfonyl fluoride, 0.1 mmol/l
aprotinin, 1 mmol/l leupeptin, 1% Triton X-100 and 10% glycerol)
was used to lyse the cell samples. The protein concentration was
estimated using a BCA protein assay kit (Pierce Biotechnology,
Rockford, IL, USA). Protein samples (30 mg) were loaded and
separated on a 10% SDS-polyacrylamide gel and then transferred
electrophoretically to nitrocellulose membranes (Pall Corporation,
East Hill, NY, USA). Nonspecific binding sites were blocked using
5% (w/v) non-fat dry milk in Tris-buffered saline with 0.1%
Tween-20 (TBST) buffer solution (0.138 mol/l NaCl, 20 mmol/l
Tris-HCl, pH 7.6, 0.1% [v/v] Tween-20; Gen-View Scientific, Inc.)
at room temperature for 1 h. Then, the membranes were incubated
overnight at 4°C with rabbit anti-p-AMPKα-Thr172 (1:1,000; cat. no.
2531) and mouse anti-GAPDH (1:3,000; cat. no. sc-32233) monoclonal
antibodies. After washing three times with TBST, the membranes were
incubated with horseradish peroxidase-conjugated goat anti-rabbit
IgG (1:6,000; cat. no. sc-2004; Santa Cruz Biotechnology, Inc.) and
goat anti-mouse IgG (1:6,000; cat. no. sc-2005; Santa Cruz
Biotechnology, Inc.) for 1 h at room temperature. An enhanced
chemiluminescence detection kit (cat. no. 32106; Pierce
Biotechnology, Inc., Rockford, IL, USA) was used to visualize the
immunoreactive bands on the membranes, according to the
manufacturer's protocol. The chemiluminescence signal was detected
by exposure to X-ray film (Kodak, Rochester, NY, USA). Densitometry
analysis was used to quantify the protein bands by calculating the
band density using Scion Image software, version 4.03 (Scion
Corporation, Frederick, MD, USA). All densitometry data are
expressed as fold-change from the control.
Statistical analysis
Values are expressed as the mean ± standard error of
the mean. The statistical significance of differences was
determined using one-way analysis of variance for multiple
comparisons with Tukey's post hoc test for analysis between groups.
P<0.05 was considered significant.
Results
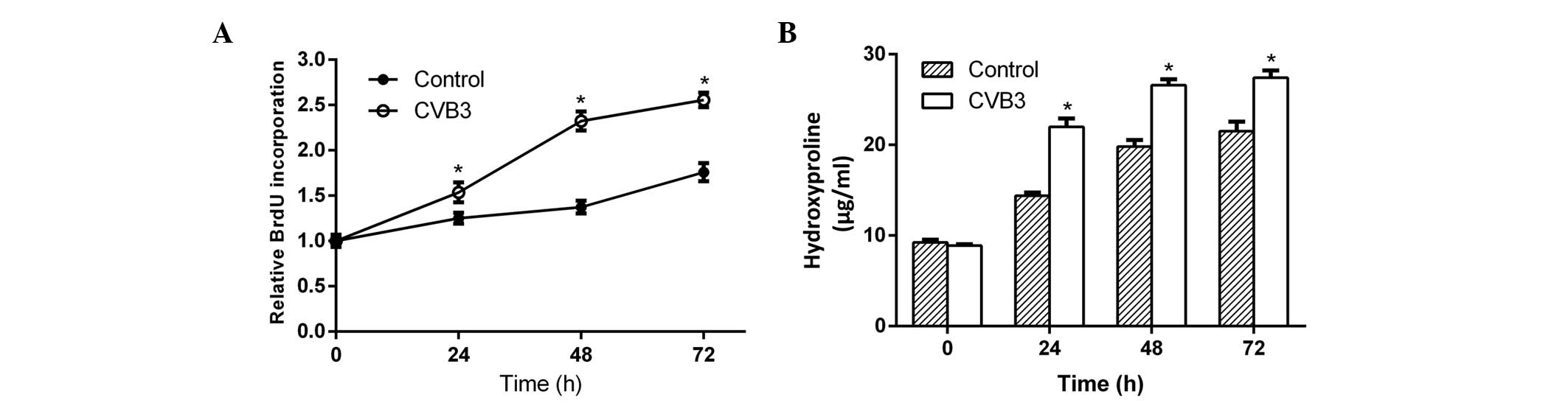
CVB3 promotes cardiac fibroblast
proliferation and collagen (hydroxyproline) secretion
To determine whether CVB3 promoted cardiac
fibroblast proliferation and collagen secretion, neonatal rat
cardiac fibroblasts incubated in serum-free DMEM medium were
infected with CVB3 (100 TCID50) for 24, 48 and 72 h. Then, newly
synthesized DNA in replicating cells and hydroxyproline content in
the culture supernatant were measured. After 24 h of infection,
both the newly synthesized DNA (Fig.
1A) and hydroxyproline (Fig. 1B)
increased significantly in the samples, and reached a peak at 48 h
after infection. This phenomenon indicated that CVB3 induced
cardiac fibroblast proliferation and collagen secretion.
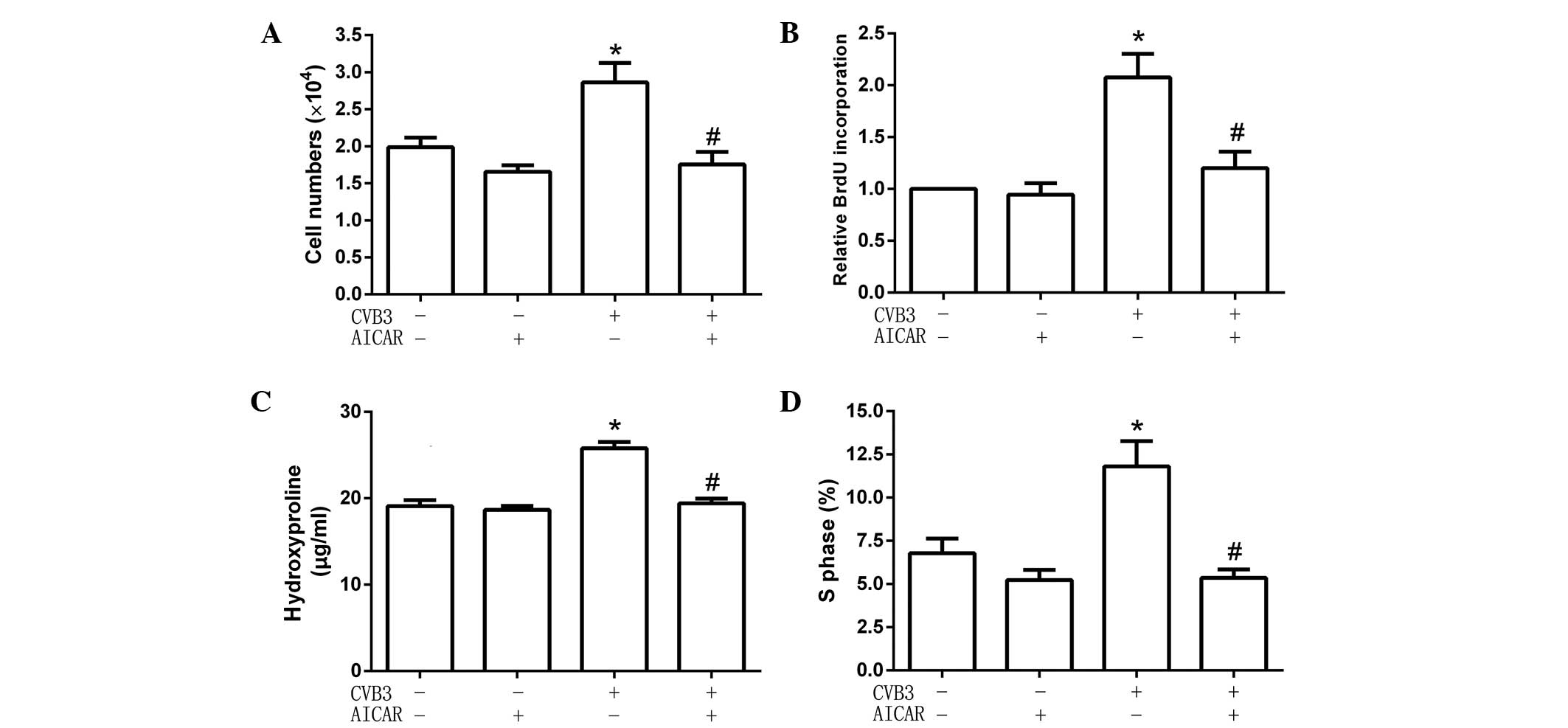
AICAR inhibits cardiac fibroblast
proliferation and collagen secretion induced by CVB3
To explore the effect of activated AMPK on cardiac
fibroblast proliferation and collagen secretion, cells were
pretreated with the specific AMPK activator AICAR (1 mmol/l) for 2
h, then infected with CVB3 (100 TCID50) for 48 h. Cell numbers, DNA
synthesis, cell cycles and hydroxyproline content were detected.
The results showed that, following pretreatment with AICAR, CVB3
did not increase cell numbers (Fig.
2A), the quantity of newly synthesized DNA in cells (Fig. 2B), hydroxyproline content in the
supernatant (Fig. 2C) and the
proportion of cells in the S phase (9.54±1.22 for cells treated
with AICAR alone vs. 5.65±0.95% for AICAR-pretreated CVB3-infected
cells; Fig. 2D). These data showed
that AICAR inhibited CVB3-induced proliferation in cardiac
fibroblasts.
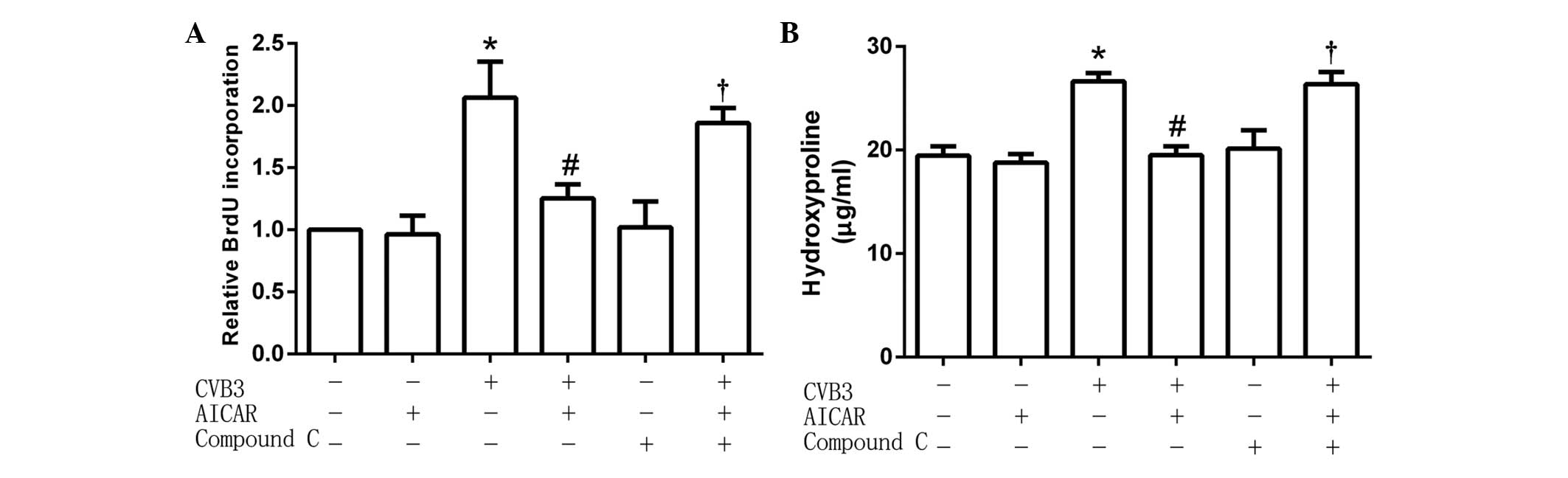
Inhibitive effects of AICAR are
attenuated by compound C
The cardiac fibroblasts (5×104 per well)
were preincubated with the specific AMPK inhibitor Compound C (1
µmol/l) for 30 min, then treated with AICAR (1 mmol/l) followed by
CVB3 (100 TICD50). In treated cells, the activity of CVB3 was
restored to increase the quantity of newly synthesized DNA in cells
(Fig. 3A) and hydroxyproline content
in the cell culture supernatant (Fig.
3B). These results suggest that the inhibitive effects of AICAR
were attenuated by pretreatment with Compound C.
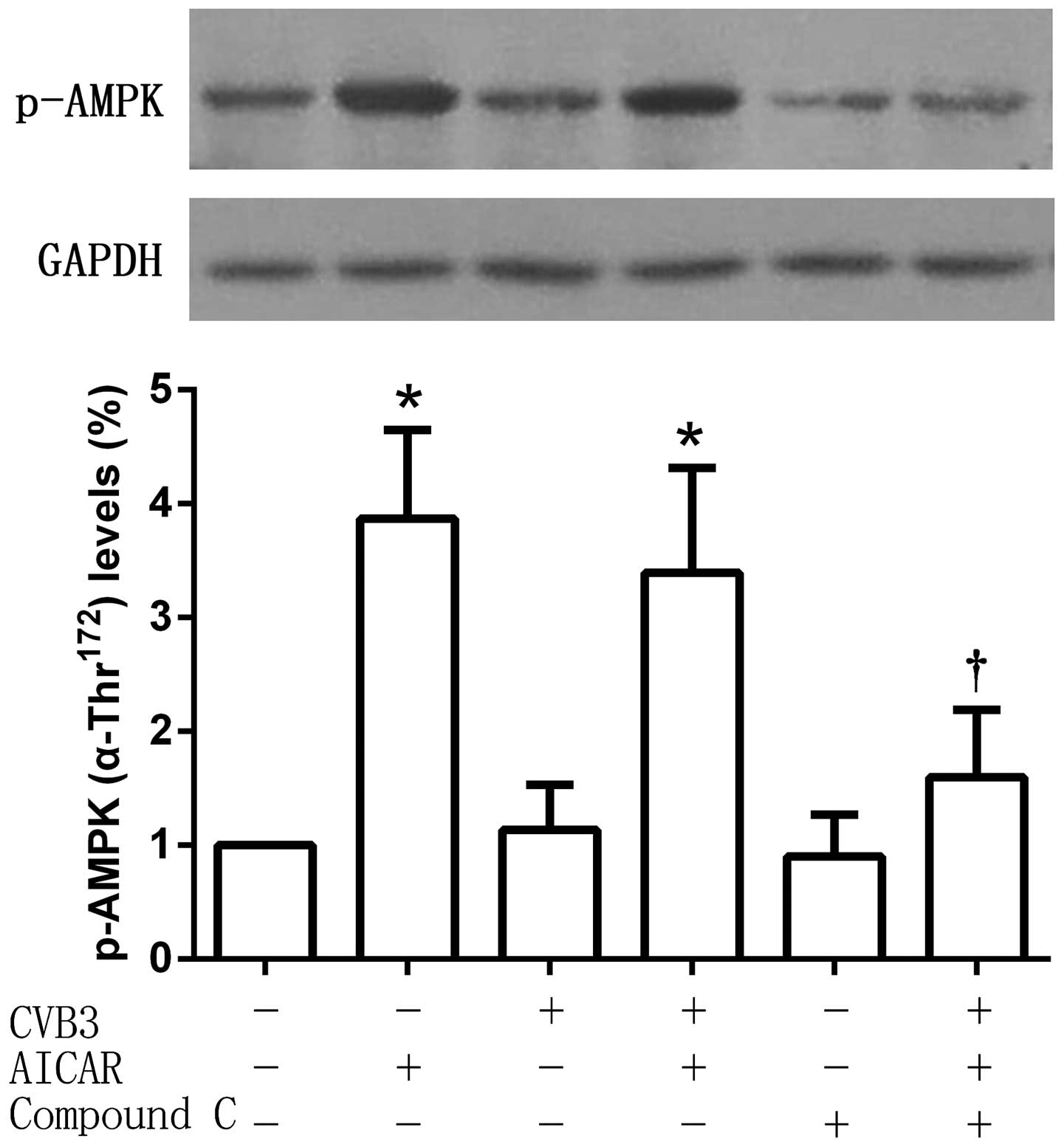
Phosphorylation of AMPKα-Thr172 is
increased by AICAR
Phospho-AMPKα-Thr172 (which serves as an
indicator of AMPK activity) protein expression levels were detected
after treatment. As an AMPK activator, AICAR significantly
increased the phosphorylation of AMPKα-Thr172, an effect
that was reversed by Compound C. No significant changes of
AMPKα-Thr172 phosphorylation were observed in the
CVB3-infected cell group compared with the untreated control, or
between the CVB3 and AICAR-treated group and the AICAR-treated
group (Fig. 4).
Discussion
CVB3 is the most common causative agent of
myocarditis (12). In the later
stage of myocarditis, persistent viral infection with CVB3 can
induce the accumulation of connective tissue and extracellular
matrix, chronic fibroblast activation and myocardial fibrosis
(13,14). CVB3 is considered to be one of the
most common causes of dilated cardiomyopathy (15). Cardiac fibroblasts, which synthesize
extracellular matrix and collagen, play a critical role in cardiac
inflammation and remodeling. Compared to cardiomyocytes, cardiac
fibroblasts aggravate viral myocarditis induced by CVB3 because of
a higher virus replication (5). They
are involved in the pathology of CVB3-induced myocarditis and
dilated cardiomyopathy (5). As a
major component of the protein collagen, hydroxyproline has an
essential role in stabilizing the triple-helical structure of
collagen, due to its effect of maximizing interchain hydrogen bond
formation (16). The measurement of
hydroxyproline levels can be used as an indicator of collagen
content (17). In the present study,
cell numbers, DNA synthesis, cell cycles and hydroxyproline content
were investigated to identify the changes occurring in cardiac
fibroblasts following CVB3 infection. The results confirmed that
CVB3 infection promoted cardiac fibroblast proliferation and
collagen secretion. This finding may contribute to our
understanding of the progression from viral myocarditis to dilated
cardiomyopathy.
AMPK is a heterotrimeric complex consisting of a
catalytic α-subunit and two regulatory subunits, namely β and γ.
The α-subunit containing a serine-threonine kinase domain has a
critical activating residue within the catalytic cleft
(Thr172) (18,19). Phosphorylation of this amino acid is
essential for AMPK activity, and its phosphorylation status often
is used as an indicator of the activation state of the kinase
(20). As a sensor of energy status,
AMPK can switch on catabolic pathways and switch off ATP-consuming
processes in order to maintain cellular energy homeostasis when
activated by metabolic stress (7).
Recent studies have elucidated that activation of the intrinsic
AMPK pathway plays an important role in the myocardial response to
ischemia (20,21), pressure overload (22) and heart failure (23). Although the mechanisms remain poorly
understood, the pleiotropic effects of AMPK in the heart are
essential to the cardioprotective actions. The compound A769662,
which enhances ischemic AMPK activation, has been found to reduce
infarct size in diabetic rat hearts (24,25).
Treatment with the AMPK activator AICAR has been demonstrated to
decrease left ventricular hypertrophy induced by aortic banding in
rats, although the treatment with AICAR also caused a reduction in
blood pressure (26). Furthermore,
metformin has been confirmed to improve the survival of patients
with heart failure and type 2 diabetes by pharmacological
activation of AMPK (27). Although
many experiments have demonstrated that AMPK activation has
pleiotropic effects in the heart, it remains unknown whether AMPK
activation is involved in the viral myocarditis induced by
CVB3.
In the present study, cell numbers, newly
synthesized DNA and the proportion of cells in the S phase were
significantly inhibited in the cardiac fibroblasts infected with
CVB3 when the cells were pretreated with AICAR. Hydroxyproline
content in the supernatant decreased, and the phosphorylation of
AMPKα-Thr172 was increased. However, these effects were
reversed following preincubation with Compound C. The results
suggest that pharmacological activation of AMPK could inhibit both
cell proliferation and collagen secretion in cardiac fibroblasts
infected by CVB3. Cardiac fibroblasts and collagen secretion are
the key causes of myocardial fibrosis, which are characterized by
excessive synthesis and accumulation of extracellular matrix
proteins (28). In the later stage
of CVB3-induced chronic myocarditis, cardiac fibrosis is an
important pathogenic factor contributing to serious cardiovascular
diseases by impairing ventricular contractility and functionality
(3). The inhibitive effects of AMPK
activation in CVB3-infected cardiac fibroblasts proliferation may
be useful in the development of effective therapies for
CVB3-induced myocarditis and dilated cardiomyopathy.
In conclusion, to the best of our knowledge, this is
the first report of an inhibitive role of pharmacological AMPK
activation in CVB3-induced cardiac fibroblast proliferation. This
finding may be helpful for the future design of therapeutic
approaches for treating cardiac fibrosis caused by chronic viral
infection, such as CVB3-induced myocarditis.
Acknowledgements
This study was supported by the Natural Science
Foundation of China (81200161) and Wuxi Hospital Management Center
Project (YGZXM14012).
References
|
1
|
Massilamany C, Huber SA, Cunningham MW and
Reddy J: Relevance of molecular mimicry in the mediation of
infectious myocarditis. J Cardiovasc Transl Res. 7:165–171. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Antoniak S and Mackman N: Coagulation,
protease-activated receptors, and viral myocarditis. J Cardiovasc
Transl Res. 7:203–211. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cao Y, Xu W and Xiong S: Adoptive transfer
of regulatory T cells protects against Coxsackievirus B3-induced
cardiac fibrosis. PLoS One. 8:e749552013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shen Y, Xu W, Chu YW, Wang Y, Liu QS and
Xiong SD: Coxsackievirus group B type 3 infection upregulates
expression of monocyte chemoattractant protein 1 in cardiac
myocytes, which leads to enhanced migration of mononuclear cells in
viral myocarditis. J Virol. 78:12548–12556. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lindner D, Li J, Savvatis K, Klingel K,
Blankenberg S, Tschöpe C and Westermann D: Cardiac fibroblasts
aggravate viral myocarditis: Cell specific coxsackievirus B3
replication. Mediators Inflamm. 2014:5195282014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brown RD, Ambler SK, Mitchell MD and Long
CS: The cardiac fibroblast: Therapeutic target in myocardial
remodeling and failure. Annu Rev Pharmacol Toxicol. 45:657–687.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hardie DG: AMPK: Positive and negative
regulation and its role in whole-body energy homeostasis. Curr Opin
Cell Biol. 33:1–7. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stuck BJ, Lenski M, Böhm M and Laufs U:
Metabolic switch and hypertrophy of cardiomyocytes following
treatment with angiotensin II are prevented by AMP-activated
protein kinase. J Biol Chem. 283:32562–32569. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang CX, Pan SN, Meng RS, Peng CQ, Xiong
ZJ, Chen BL, Chen GQ, Yao FJ, Chen YL, Ma YD and Dong YG: Metformin
attenuates ventricular hypertrophy by activating the AMP-activated
protein kinase-endothelial nitric oxide synthase pathway in rats.
Clin Exp Pharmacol Physiol. 38:55–62. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xie W, Wang L, Dai Q, Yu H, He X, Xiong J,
Sheng H, Zhang D, Xin R, Qi Y, et al: Activation of AMPK restricts
coxsackievirus B3 replication by inhibiting lipid accumulation. J
Mol Cell Cardiol. 85:155–167. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Reed LJ and Muench H: A simple method of
estimating fifty percent endpoints. Am J Hyg. 27:493–497. 1938.
|
|
12
|
Liu MY, Wu DL, Liu NH, Meng QW and Meng
FC: 1A and 3D gene sequences of coxsackievirus B3 strain CC:
Variation and phylogenetic analysis. DNA Seq. 19:8–12. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Leipner C, Grün K, Schneider I, Glück B,
Sigusch HH and Stelzner A: Coxsackievirus B3-induced myocarditis:
Differences in the immune response of C57BL/6 and Balb/c mice. Med
Microbiol Immunol. 193:141–147. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chapman NM and Kim KS: Persistent
coxsackievirus infection: Enterovirus persistence in chronic
myocarditis and dilated cardiomyopathy. Curr Top Microbiol Immunol.
323:275–292. 2008.PubMed/NCBI
|
|
15
|
Pankuweit S and Klingel K: Viral
myocarditis: From experimental models to molecular diagnosis in
patients. Heart Fail Rev. 18:683–702. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bhattacharjee A and Bansal M: Collagen
structure: The Madras triple helix and the current scenario. IUBMB
Life. 57:161–172. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kasyanov V, Moreno-Rodriguez RA, Kalejs M,
Ozolanta I, Stradins P, Wen X, Yao H and Mironov V: Age-related
analysis of structural, biochemical and mechanical properties of
the porcine mitral heart valve leaflets. Connect Tissue Res.
54:394–402. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu WY and Jiang RS: Advances in the
research of AMPK and its subunit genes. Pak J Biol Sci.
16:1459–1468. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xiao B, Sanders MJ, Underwood E, Heath R,
Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, et
al: Structure of mammalian AMPK and its regulation by ADP. Nature.
472:230–233. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Baron SJ, Li J, Russell RR III, Neumann D,
Miller EJ, Tuerk R, Wallimann T, Hurley RL, Witters LA and Young
LH: Dual mechanisms regulating AMPK kinase action in the ischemic
heart. Circ Res. 96:337–345. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ma Y, Wang J, Gao J, Yang H, Wang Y,
Manithody C, Li J and Rezaie AR: Antithrombin up-regulates
AMP-activated protein kinase signalling during myocardial
ischaemia/reperfusion injury. Thromb Haemost. 113:338–349. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li Y, Chen C, Yao F, Su Q, Liu D, Xue R,
Dai G, Fang R, Zeng J, Chen Y, et al: AMPK inhibits cardiac
hypertrophy by promoting autophagy via mTORC1. Arch Biochem
Biophys. 558:79–86. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Beauloye C, Bertrand L, Horman S and Hue
L: AMPK activation, a preventive therapeutic target in the
transition from cardiac injury to heart failure. Cardiovasc Res.
90:224–233. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim AS, Miller EJ, Wright TM, Li J, Qi D,
Atsina K, Zaha V, Sakamoto K and Young LH: A small molecule AMPK
activator protects the heart against ischemia-reperfusion injury. J
Mol Cell Cardiol. 51:24–32. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Paiva MA, Rutter-Locher Z, Gonçalves LM,
Providência LA, Davidson SM, Yellon DM and Mocanu MM: Enhancing
AMPK activation during ischemia protects the diabetic heart against
reperfusion injury. Am J Physiol Heart Circ Physiol.
300:H2123–H2134. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li HL, Yin R, Chen D, Liu D, Wang D, Yang
Q and Dong YG: Long-term activation of adenosine
monophosphate-activated protein kinase attenuates
pressure-overload-induced cardiac hypertrophy. J Cell Biochem.
100:1086–1099. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gundewar S, Calvert JW, Jha S,
Toedt-Pingel I, Ji SY, Nunez D, Ramachandran A, Anaya-Cisneros M,
Tian R and Lefer DJ: Activation of AMP-activated protein kinase by
metformin improves left ventricular function and survival in heart
failure. Circ Res. 104:403–411. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yamazaki KG, Gonzalez E and Zambon AC:
Crosstalk between the renin-angiotensin system and the advance
glycation end product axis in the heart: Role of the cardiac
fibroblast. J Cardiovasc Transl Res. 5:805–813. 2012. View Article : Google Scholar : PubMed/NCBI
|