Introduction
Laryngeal squamous cell carcinomas (LSCCs) account
for >85% of all malignant tumors occurring in the larynx
(1), and despite the great progress
that has been made in the treatment of the disease, including
surgery or radiotherapy (2,3), the molecular basis of LSCC remains
unclear.
Numerous studies have shown that microRNAs (miRNAs),
a class of small, single-stranded, non-coding RNAs, play a critical
role in the regulation of tumorigenesis (4,5). The
aberrant expression of certain miRNAs has been shown to regulate
cell proliferation, apoptosis, invasion and angiogenesis in human
malignancies, either through the induction of mRNA degradation, or
through translational inhibition (5,6).
microRNA-153 (miR-153) is located in intron 19 of
genes encoding two of the major type 1 diabetes autoantigens,
islet-associated protein (IA)-2 and IA-2β (7). Kim et al found that miR-153 was
downregulated and correlated with advanced clinical stage in
ovarian epithelial tumors (8). In
addition, the downregulation of miR-153 has been found to
contribute to epithelial-mesenchymal transition and tumor
metastasis in human epithelial cancer (9). Furthermore, the levels of miR-153 have
been found to be decreased in LSCC tissues compared with the
adjacent tissues (10). In cultured
lung cancer cells, the overexpression of miR-153 has been shown to
significantly inhibit cell proliferation and migration, and promote
apoptosis in through the suppression of protein kinase B (AKT1)
(10); however, miR-153 has been
observed to be upregulated in human colorectal cancer (CC) cells,
thereby supporting CC progression and chemotherapeutic resistance
(11). Based on the aforementioned
results, the role of miR-153 in human cancers may be cell- or
tissue-specific.
In the present study, the expression of miR-153 in
LSCC tissues was evaluated, and the effect of miR-153 transfection
on human epithelial cells was evaulated in vitro, with the
aim of elucidating the role and mechanisms of miR-153 in laryngeal
tumorigenesis.
Materials and methods
Cinical tissue samples
LSCC tissue and adjacent control mucosa specimens
were collected from 38 patients (age, 22–54 years; males, 22;
females, 16) who had undergone total or partial laryngectomy
between May 2011 and December 2013. Informed consent was obtained
from each patient prior to sample collection. The present study was
reviewed and approved by the Ethics Committee of Zhangjiagang First
People's Hospital (Suzhou, China).
Cell culture and transfection
Human epithelial type 2 (HEp-2) cells were purchased
from American Type Culture Collection (Rockville, MD, USA). Cells
were cultured in RPMI-1640 medium (Gibco; Thermo Fisher Scientific,
Inc., Shanghai, China) supplemented with 10% fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Inc.). The cultured cells
were incubated at 37°C with 5% CO2 in a humidified
incubator. Human miR-153 mimics (miR-153-1), antisense
oligonucleotide (ASO) and negative controls (NCs) were obtained
from Shanghai GenePharma Co., Ltd. (Shanghai, China). For
transfection, cells were cultured in a 6-well plate and transiently
transfected at 70–80% confluence using Lipofectamine 2000 reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) following the
manufacturer's protocol.
Cell viability assay
The viability of the cells was determined by
assaying the reduction of
3-(4,5-dimethylthiazol-2-yl)-2,5-di-phenyltetrazolium bromide (MTT;
Beyotime Institute of Biotechnology Co., Shanghai, China) to
formazan, following the manufacturer's protocol.
Cell proliferation, cell cycle, and
invasion assays
Cell proliferation was evaluated by analyzing the
incorporation of bromodeoxyuridine (BrdU) into cellular DNA during
DNA synthesis using an enzyme-linked immunosorbent assay (BrdU kit;
Beyotime Institute of Biotechnology Co., Shanghai, China),
following the manufacturer's instructions. All experiments were
performed in triplicate. Absorbance was measured using the Spectra
Max 190 ELISA reader (Molecular Devices, Sunnyvale, CA, USA) at 450
nm. For cell-cycle analysis, cells were labeled for 20 min with
propidium iodide (Beyotime Institute of Biotechnology Co.) and
immediately analyzed using Gallios™ flow cytometry (Beckman
Coulter, Inc., Brea, CA, USA). A total of 10,000 cells were
acquired and analyzed for DNA content. All data were collected,
stored, and analyzed using ModFit 3.2 software (Verity Software
House Company, Topsham, ME, USA). Histograms represent the
percentage of cells in each phase of the cell cycle (G0/G1, S and
G2/M).
Cell invasion assays were performed using
extracellular matrix-coated invasion chambers (ECM550; EMD
Millipore, Temecula, CA, USA), according to the manufacturer's
instructions. Briefly, cells were harvested and resuspended in
serum-free medium following pretreatment for 24 h with miR-153
mimic/miR-153 antisense or negative control. A total of
2×105 cells were plated into the top well of an
extracellular matrix-coated invasion chamber, whereas the bottom
well of the chamber contained 500 µl Dulbecco's Modified Eagle's
Medium supplemented with 10% FBS. After 24-h incubation, the
noninvading cells were removed using a cotton swab and the invading
cells on the underside of the membrane were stained with Giemsa
cell stain solution (Millipore) for 10 min. Following washing three
times with water, 100 µl extraction buffer (EMD Millipore) was used
to remove the stain from each membrane and quantified using a
colorimetric microplate reader at 570 nm (Bio-Rad Laboratories,
Shanghai, China), according to the manufacturer's instructions.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA from fresh tissues and cultured cells was
harvested using an mirVana miRNA Isolation kit (Ambion, Austin, TX,
USA) according to the manufacturer's protocol. A NanoDrop ND-1000
spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) was
used to determine the RNA concentration. RNA was treated with DNase
(PureLink DNase Set; 12185010; Thermo Fisher Scientific, Inc.)
prior to analysis. RT-qPCR was performed using TaqMan Universal PCR
Master mix (Applied Biosystems; Thermo Fisher Scientific, Inc.) and
an ABI Prism 7900HT Real-Time PCR System (Thermo Fisher Scientific,
Inc.). The primer for miR-153 was GATCACTTTTGTGACTATGCAACT. Small
nuclear U6 snRNA (Applied Biosystems) was used as an internal
control for the expression of miR-153.
To analyze the relative mRNA levels of cyclin B1 and
cyclin D1, total RNA was isolated using TRIzol reagent according to
the manufacturer's protocol (Invitrogen; Thermo Fisher Scientific,
Inc.). First-strand complementary DNA (cDNA) synthesis was
conducted for each RNA sample using the Promega Reverse
Transcription System (Promega Corporation, Madison, WI, USA). Oligo
dT was employed to prime cDNA synthesis. qPCR was performed using
Takara SYBR Master mix (Takara Bio., Inc., Shiga, Japan) and a
LightCycler 480 instrument (Roche Diagnostics, Basel, Switzerland).
PCR conditions included an initial holding period at 95°C for 5 sec
and 60°C for 30 sec for 45 cycles. The primer sequences were:
Cyclin B1, forward: 5′-AATAAGGCGAAGATCAACATGGC-3′, and reverse:
5′-TTTGTTACCAATGTCCC CAAGAG-3′; cyclin D1, forward:
5′-TGGAGCCCGTGAAAAAGAGC-3′, and reverse:
5′-TCTCCTTCATCTTAGAGGCCAC-3′). Differences in gene expression were
calculated using the 2−ΔΔCq method (12) and expressed as a fold-change. Each
experiment was repeated ≥3 times.
Western blot analysis
Cells or tissues were harvested and lysed with lysis
buffer (150 mm NaCl, 50 mm Tris-HCl, 1% NP-40, pH 7.5; Beyotime
Institute of Biotechnology Co.). Proteins were quantified and
separated in 8% sodium dodecyl sulfate-agarose gel and then
transferred to nitrocellulose membranes (Amersham Biosciences,
Little Chalfont, Buckinghamshire, UK). Following blocking with 10%
nonfat milk in phosphate-buffered saline, membranes were
immunoblotted with primary antibodies as listed below. The
membranes were incubated with primary antibodies at 4°C overnight
and washed three times in 0.1% Tris-buffered saline and Tween 20
(TBST; Beyotime Institute of Biotechnology Co.). They were then
incubated with mouse anti-rabbit (1:1,500; sc-2491; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) and rabbit anti-mouse IgG
(1:2,000; ab193651; Abcam, Cambridge, MA, USA) secondary antibodies
for 2 h at 25°C and washed three times in 0.1% TBST. They were then
incubated with secondary antibodies (Santa Cruz Company,
California, USA) for 2 hr at 25℃ and washed three times each in 0.1
%TBST. The signals were detected using SuperSignal West Pico
Chemiluminescent Substrate kit (Pierce Biotechnology, Inc.,
Rockford, IL, USA) according to the manufacturer's instructions.
The primary antibodies anti-cyclin B1 (mouse monoclonal antibody;
ab72; 1:3,000), anti-cyclin D1 (rabbit polyclonal antibody;
ab16663; 1:2,000), anti-kruppel-like factor 5 (KLF5; rabbit
polyclonal antibody; ab24331; 1:2,000), anti-AKT1 (rabbit
polyclonal antibody; ab32505; 1:1,000), anti-forkhead box O3
(FOXO3a; rabbit polyclonal antibody; ab12162; 1:1,000) and
anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; rabbit
polyclonal antibody; ab181602, 1:1,000) were purchased from Abcam.
Protein levels were normalized to total GAPDH expression.
Luciferase reporter assay
Potential target genes for miR-153 were searched for
using bioinformatics software (miRWalk) (13). The 3′-untranslated region (3′-UTR) of
the human gene KLF5 was predicted to interact with miR-153. The
3′-UTR of KLF5 was synthesized and inserted into the luciferase
reporter construct pMir-Report (Ambion; Thermo Fisher Scientific,
Inc.), yielding pMir-Report-KLF5. Mutations within potential
miR-153 binding sites were generated by nucleotide replacement of
the wild type sequence to inhibit miR-153 binding. HEp-2 cells were
transfected with either miR-153 mimics or NC, together with
pMir-Report vectors containing wild-type or mutant KLF5 3′-UTR
variants for 36 h. The pRL-SV40 vector (Promega Corporation)
carrying the Renilla luciferase gene was used as an internal
control to normalize the transfection efficiency, and the
Dual-Luciferase Reporter Assay System (Promega Corporation) was
used to determine the luciferase values.
Statistical analysis
Data were collected from at least four separate
experiments and are expressed as the mean ± standard error. The
differences between groups were analyzed using the Student's t-test
or one-way analysis of variance. P<0.05 was considered to
indicate a statistically significant difference.
Results
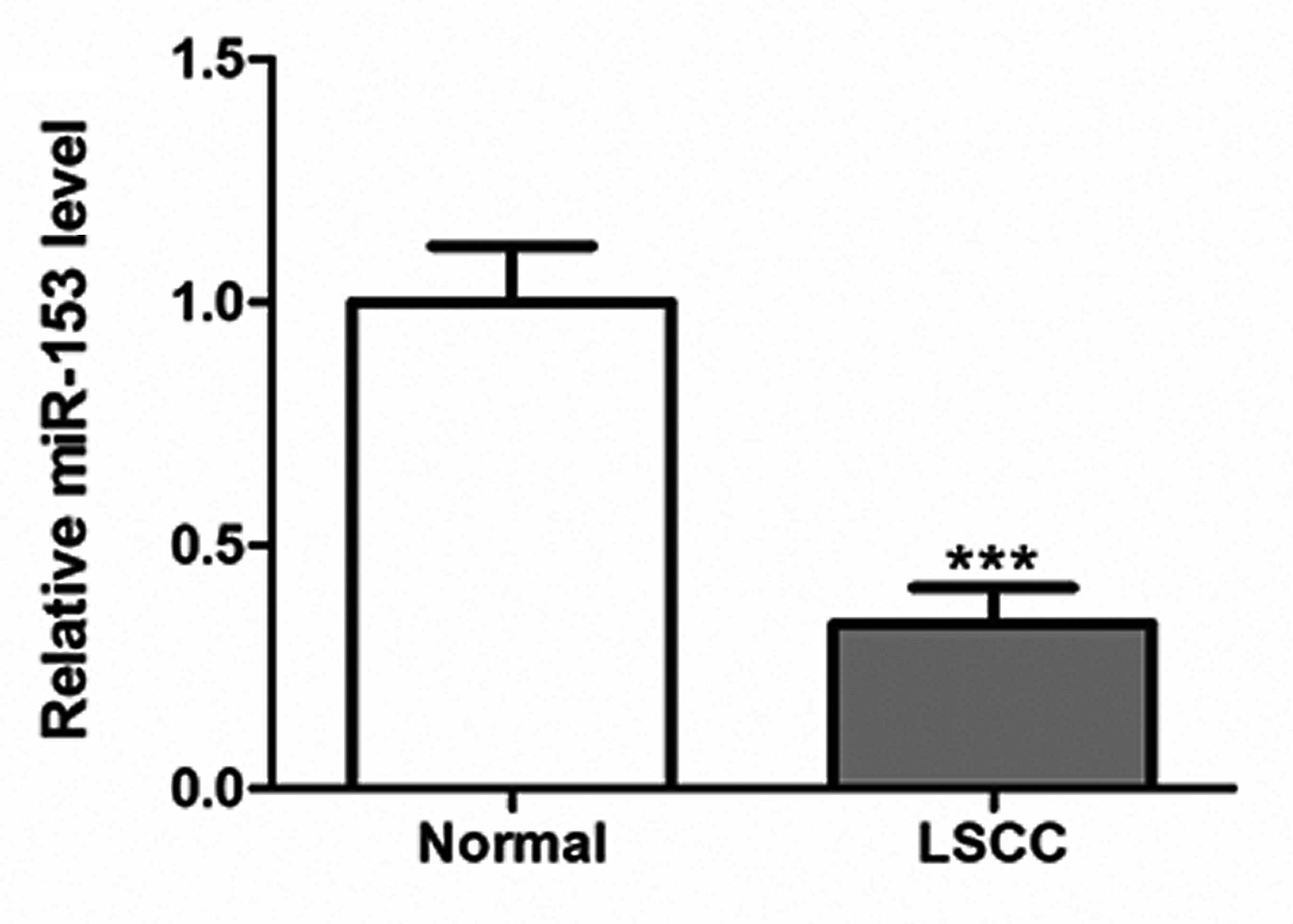
Downregulation of miR-153 in LSCC
tissues
RT-qPCR was performed to determine the expression
levels of miR-153 in LSCC tissues and control mucosa specimens.
miR-153 was found to be significantly downregulated in the LSCC
tissues compared with the control (P<0.001; Fig. 1).
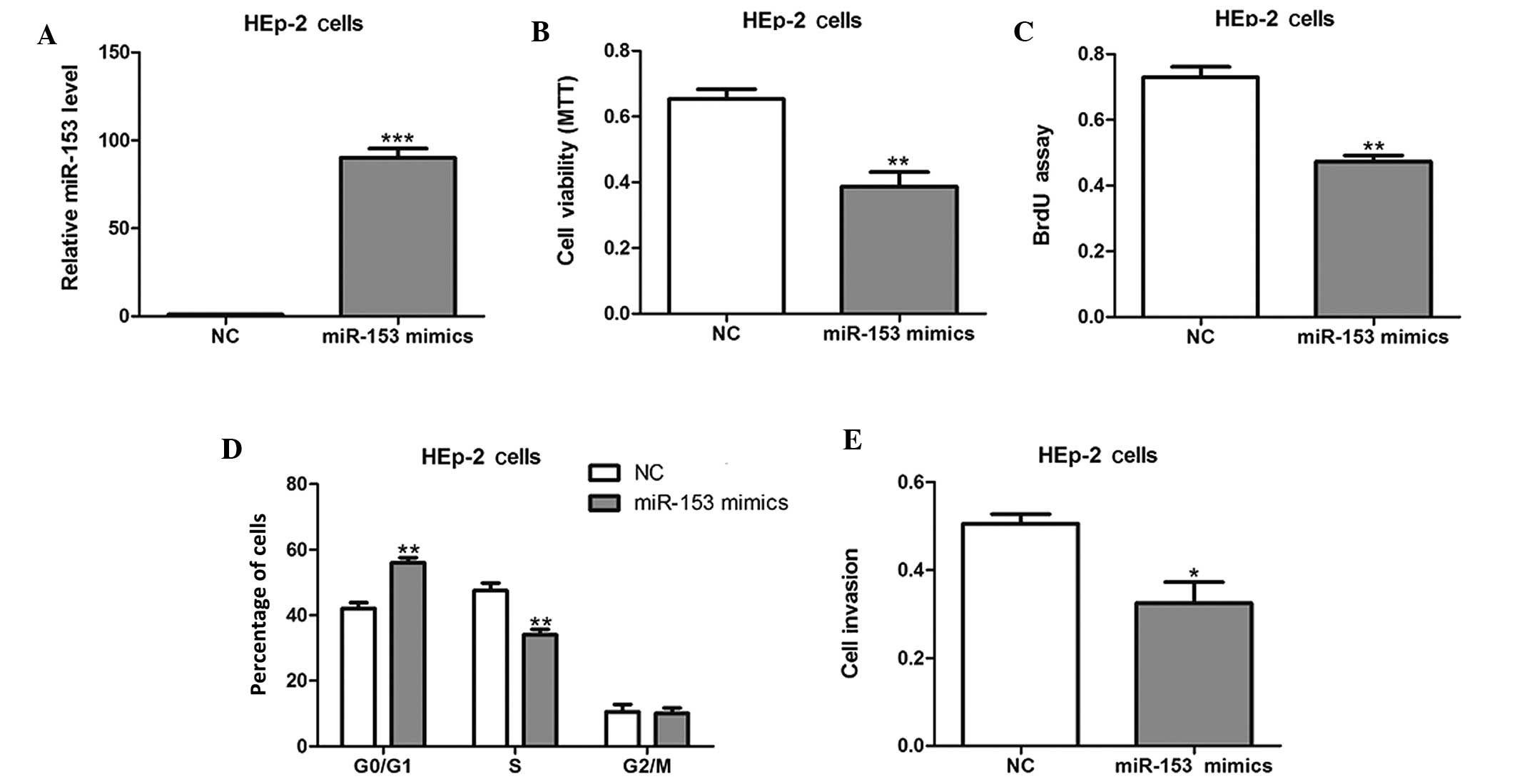
Effects of miR-153 on HEp-2 cell
proliferation and invasion
In order to evaluate the effect of miR-153 on cell
growth, HEp-2 cells were transiently transfected with miR-155
mimics or NCs (Fig. 2A). The forced
expression of miR-153 mimics markedly inhibited cell viability and
proliferation, as shown by the results of MTT and BrdU
incorporation assays (P<0.01; Fig. 2B
and C). In addition, cell cycle analysis revealed that the
percentage of miR-153 mimic-transfected cells in the G1/G0 phase
was notably higher than the percentage in the S phase (Fig. 2D). Furthermore, the invasive
abilities of the HEp-2 cells were also found to be reduced by
miR-153 mimics (Fig. 2E).
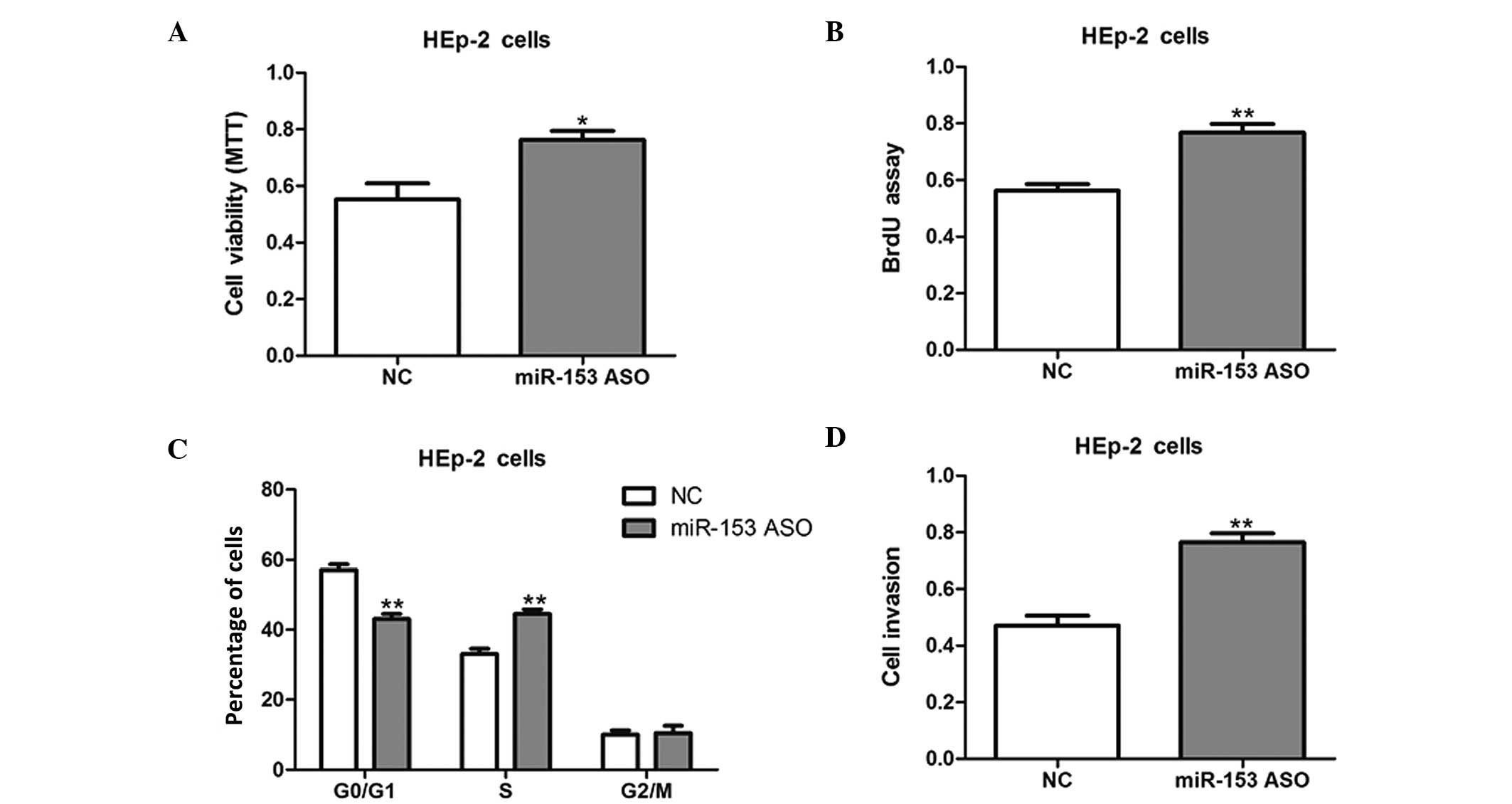
HEp-2 cells were then transfected with miR-153 ASO
to knock down miR-153 expression. The inhibition of miR-153
promoted the cell growth and invasion of HEp-2 cells, as compared
with NC-transfected cells (Fig. 3);
therefore, the present results suggest that miR-153 inhibits cell
proliferation and invasion in vitro.
miR-153 suppresses KLF5 in LSCC
cells
AKT1 and FOXO3a have been shown to be negatively
regulated by miR-153 in human cancers (10,11);
however, no changes in the protein levels of AKT1 and FOXO3a among
the HEp-2 cells transfected with miR-153 mimics, ASO or NC were
detected in the present study (Fig. 4A
and B). Potential target genes for miR-153 were therefore
searched for using bioinformatics software. It was found that KLF5
harbors a potential miR-153 binding site in its 3′-UTR (Fig. 4C). Subsequently, the 3′-UTR of the
KLF5 gene (wild type and mutant) was cloned and inserted into a
luciferase reporter construct. As shown in Fig. 4D, the overexpression of miR-153
mimics led to a reduction in luciferase activity when the
luciferase reporter construct contained the wild-type KLF5 3′-UTRs;
however, mutation of the potential miR-153 binding site abolished
the reduction in luciferase expression (Fig. 4D). Furthermore, the transfection of
miR-153 mimics in HEp-2 cells resulted in a reduction in the level
of KLF5 protein expression (Fig.
4E). Consistent with these findings, a marked upregulation of
KLF5 was observed in the HEp-2 cells with miR-153 inhibition
(Fig. 4F). The results of the
present study therefore suggest that KLF5 may be a target of
miR-153 in LSCC cells.
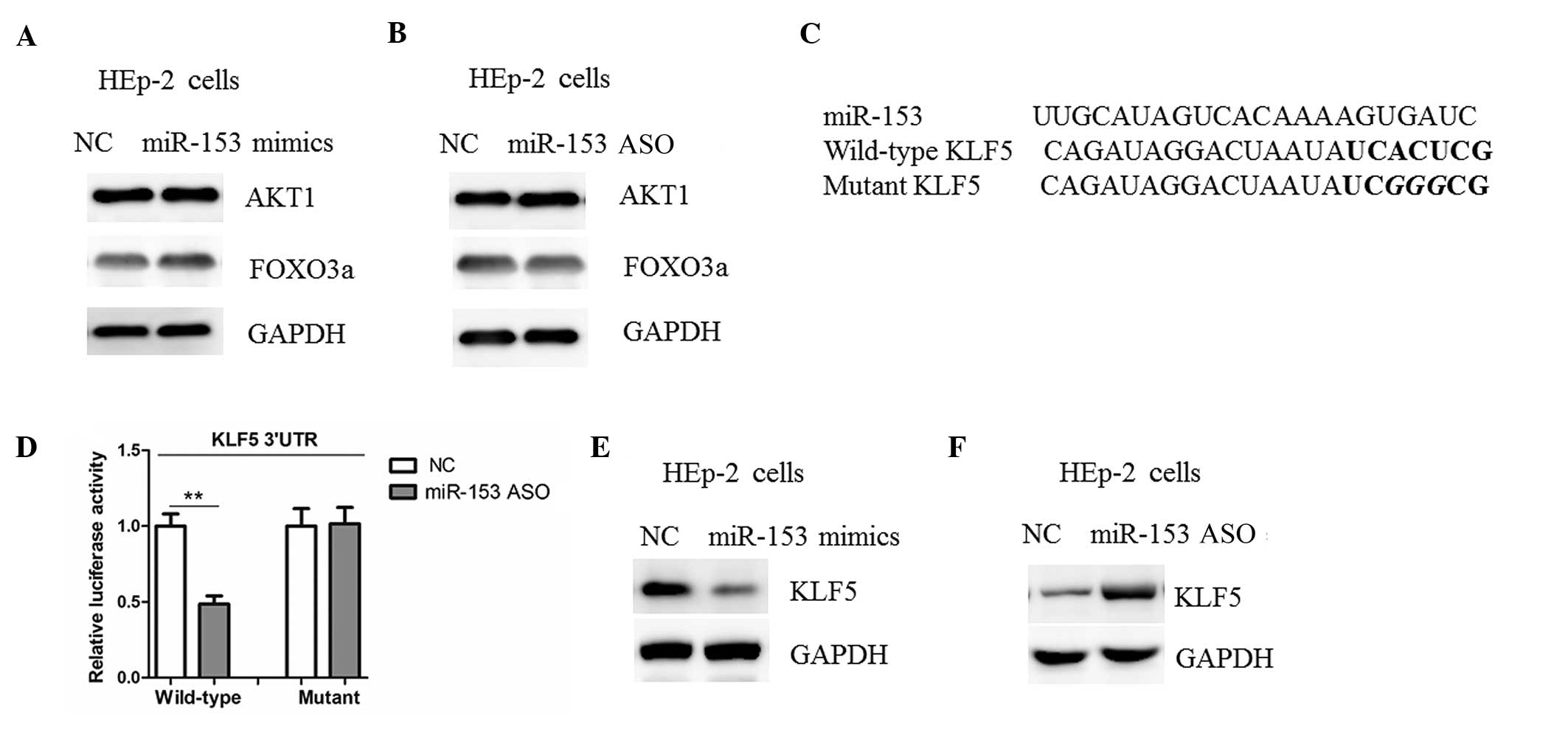 | Figure 4.miR-153 negatively regulates KLF5
expression in LSCC. (A and B) Western blot analysis of AKT1 and
FOXO3a expression in HEp-2 cells transfected with miR-153 mimics,
ASO or NC. (C) Informatics prediction of miR-153 binding sites in
the 3′-UTRs of KLF5. (D) Luciferase reporter assays in HEp-2 cells.
Cells were transfected with wild-type 3′-UTR-reporter or mutant
constructs together with miR-153 mimics or NC. Representative
western blot analysis of KLF5 expression in HEp-2 cells transfected
with (E) miR-153 mimics or NC and (F) miR-153 antisense or NC.
**P<0.01 between the two groups. KLF5, anti-kruppel-like factor
5; miR-153, microRNA-153; HEp-2, human epithelial type 2; 3′-UTR,
3′-untranslated region; NC, negative control; ASO, antisense
oligonucleotide; LSCC, laryngeal squamous cell carcinoma; AKT1,
protein kinase B; FOXO3a, forkhead box O3. |
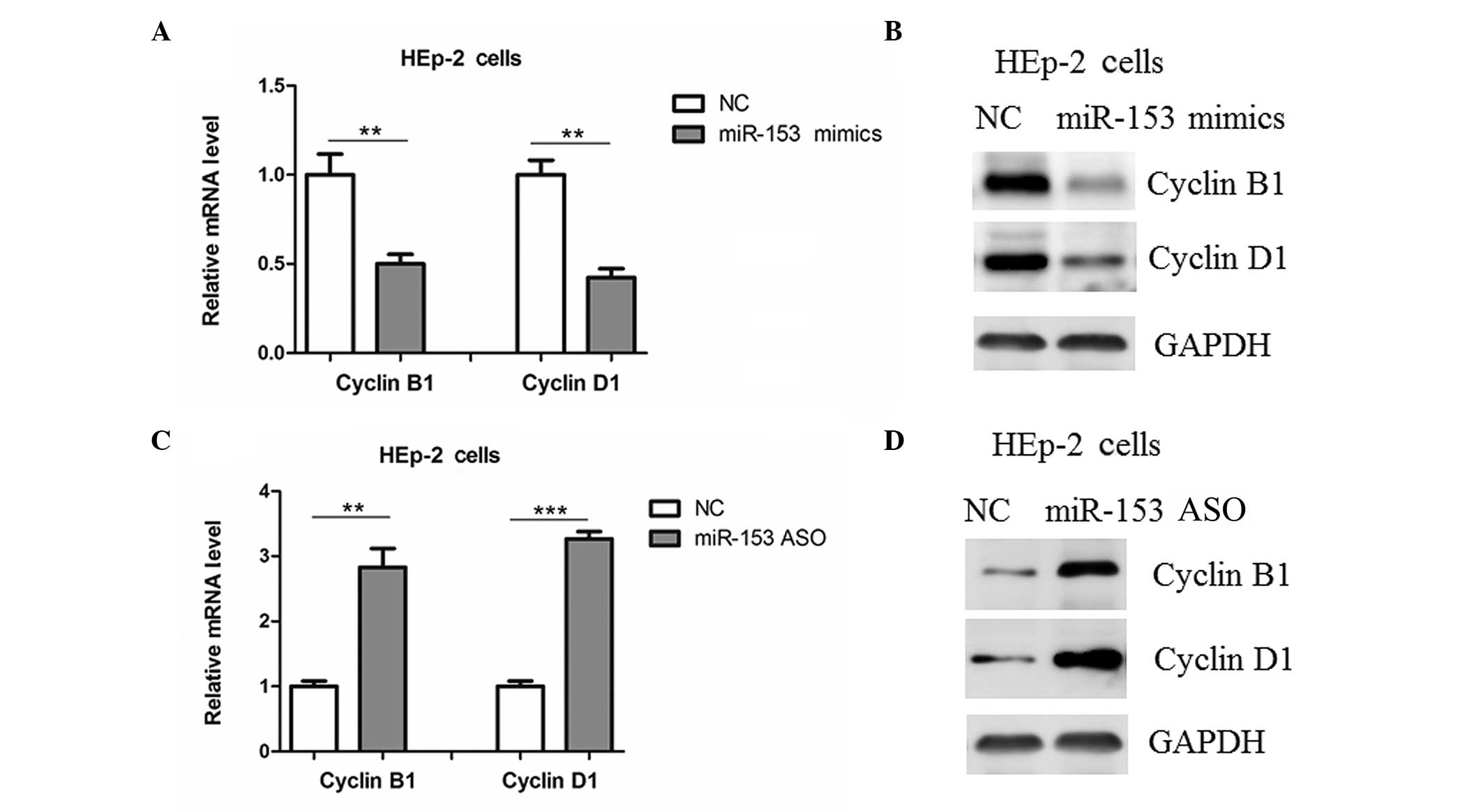
Regulation of cyclins B1 and D1 by
miR-153
Previous studies have shown that KLF5 may promote
cell proliferation through the upregulation of cell-cycle
regulators, such as cyclins B1 and D1 (14,15). In
the present study, it was found that the transfection of HEp-2
cells with miR-153 mimics reduced the mRNA and protein expression
levels of cyclin B1 and cyclin D1 (Fig.
5A and B), and the expression levels of these cyclins in HEp-2
cells were upregulated by miR-153 ASO (Fig. 5C and D).
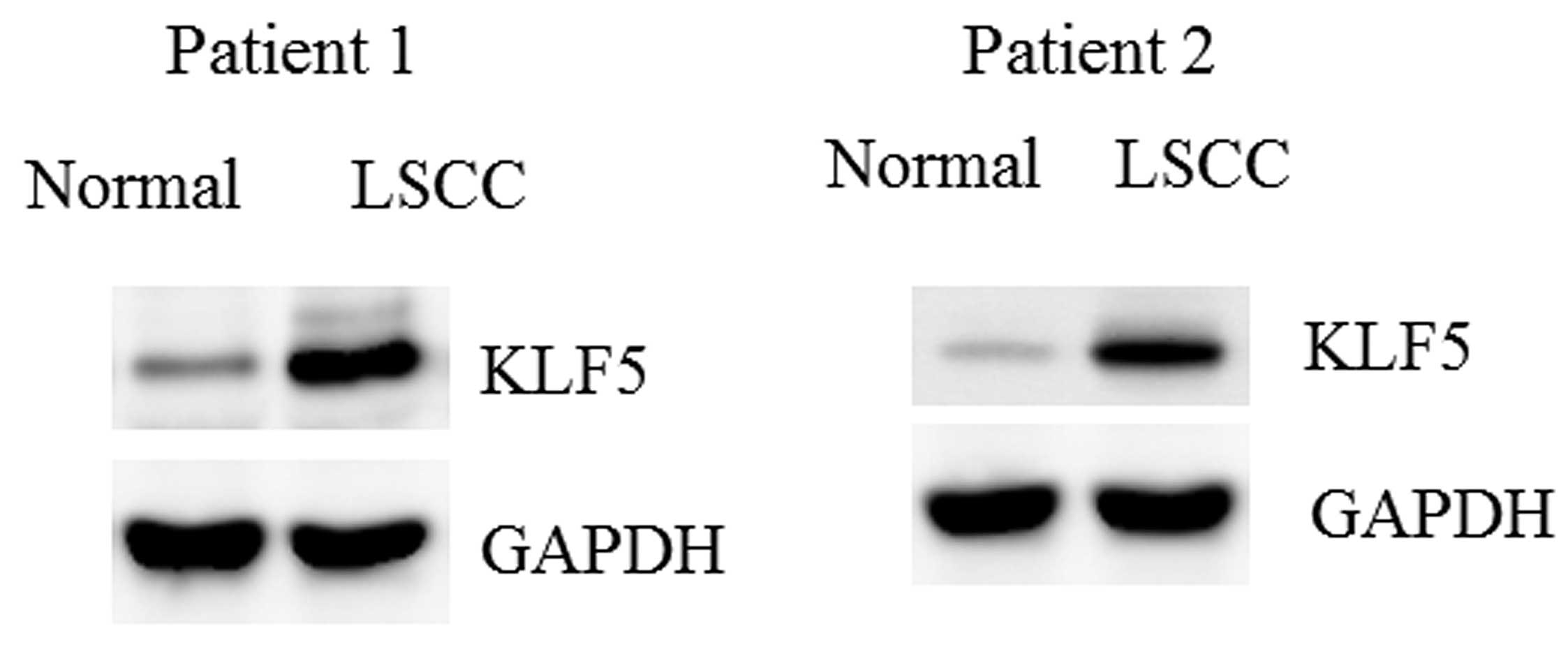
Upregulation of KLF5 in LSCC
tissues
The protein levels of KLF5 in the LSCC tissues and
control mucosa specimens were determined. Consistent with the
downregulation of miR-153, KLF5 was markedly upregulated in the
LSCC tissues (Fig. 6).
Discussion
In the present study, miR-153 expression was found
to be downregulated in LSCC tissues. In vitro experiments
further indicated that miR-153 may inhibit LSCC cell proliferation
and progression through the suppression of KLF5; however, the exact
mechanism of the inhibition of miR-153 expression in LSCC remains
unclear. Notably, a previous study has found that cytosine
methylation in miR-153 gene promoters could modulate its expression
in human kidney cells (16);
therefore, it may be speculated that epigenetic regulation might be
involved in the downregulation of miR-153 in LSCC tissues.
Previous studies have identified AKT1 and FOXO3a as
translational targets for miR-153 (10,11);
however, the present study showed that the abundance of these
proteins was not affected in HEp-2 cells transfected with miR-153
mimics or ASO. Although the reason for this inconsistency is not
known, the role of miR-153 in human cancers may be tissue- or
cell-specific. Furthermore, KLF5, a new direct and functional
target of miR-153, was identified in LSCC. miR-153 was shown to be
able to bind to the 3′-UTR of KLF5 to suppress its protein
contents. Studies have shown that KLF5 acts as an oncogene in
various types of human cancers (17,18). The
upregulation of KLF5 has been observed in breast, colon, gastric
and prostate cancers (17–21). In addition, KLF5 promotes cell
survival and inhibits apoptosis in cancer cells via multiple
mechanisms (22,23). For instance, KLF5 has been shown to
interact with tumor suppressor p53 in regulating the expression of
the inhibitor-of-apoptosis protein survivin, which may play a role
in the pathological process of cancer (24). KLF5 has also been found to promote
lung tumorigenesis through the upregulation of the Sox4 gene
(25). Despite the aforementioned
findings, KLF5 remains a poor candidate as a drug target, due to
the lack of a ligand-binding domain and broad transcriptional
signature; therefore, a better understanding of the regulatory
pathways for the manipulation of its expression could provide
impetus to the identification of pharmacological approaches to
treat human cancers, including LSCC.
In conclusion, a downregulation of miR-153 was
observed in LSCC tissues, and it was demonstrated that miR-153 may
act as a tumor suppressor in LSCC initiation and/or progression. It
was further found that miR-153 possesses the potency to suppress
LSCC growth and invasion through the suppression of KLF5,
suggesting that miR-153 could potentially serve as a therapeutic
target for LSCC.
References
|
1
|
Nadal A and Cardesa A: Molecular biology
of laryngeal squamous cell carcinoma. Virchows Arch. 442:1–7.
2003.PubMed/NCBI
|
|
2
|
Lionello M, Staffieri A and Marioni G:
Potential prognostic and therapeutic role for angiogenesis markers
in laryngeal carcinoma. Acta Otolaryngol. 132:574–582. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Caicedo-Granados E, Beswick DM,
Christopoulos A, Cunningham DE, Razfar A, Ohr JP, Heron DE and
Ferris RL: Oncologic and functional outcomes of partial laryngeal
surgery for intermediate-stage laryngeal cancer. Otolaryngol Head
Neck Surg. 148:235–242. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schwarzenbach H, Nishida N, Calin GA and
Pantel K: Clinical relevance of circulating cell-free microRNAs in
cancer. Nat Rev Clin Oncol. 11:145–156. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kasinski AL and Slack FJ: Epigenetics and
genetics. MicroRNAs en route to the clinic: Progress in validating
and targeting microRNAs for cancer therapy. Nat Rev Cancer.
11:849–864. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ryan BM, Robles AI and Harris CC: Genetic
variation in microRNA networks: The implications for cancer
research. Nat Rev Cancer. 10:389–402. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mandemakers W, Abuhatzira L, Xu H,
Caromile LA, Hébert SS, Snellinx A, Morais VA, Matta S, Cai T,
Notkins AL and De Strooper B: Co-regulation of intragenic microRNA
miR-153 and its host gene Ia-2 β: Identification of miR-153 target
genes with functions related to IA-2β in pancreas and brain.
Diabetologia. 56:1547–1556. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim TH, Kim YK, Kwon Y, Heo JH, Kang H,
Kim G and An HJ: Deregulation of miR-519a, 153 and 485-5p and its
clinicopathological relevance in ovarian epithelial tumours.
Histopathology. 57:734–743. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu Q, Sun Q, Zhang J, Yu J, Chen W and
Zhang Z: Downregulation of miR-153 contributes to
epithelial-mesenchymal transition and tumor metastasis in human
epithelial cancer. Carcinogenesis. 34:539–549. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yuan Y, Du W, Wang Y, Xu C, Wang J, Zhang
Y, Wang H, Ju J, Zhao L, Wang Z, et al: Suppression of AKT
expression by miR-153 produced anti-tumor activity in lung cancer.
Int J Cancer. 136:1333–1340. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang L, Pickard K, Jenei V, Bullock MD,
Bruce A, Mitter R, Kelly G, Paraskeva C, Strefford J, Primrose J,
et al: MiR-153 supports colorectal cancer progression via
pleiotropic effects that enhance invasion and chemotherapeutic
resistance. Cancer Res. 73:6435–6447. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Erickson HS, Albert PS, Gillespie JW,
Wallis BS, Rodriguez-Canales J, Linehan WM, Gonzalez S, Velasco A,
Chuaqui RF and Emmert-Buck MR: Assessment of normalization
strategies for quantitative RT-PCR using microdissected tissue
samples. Lab Invest. 87:951–962. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dweep H, Gretz N and Sticht C: MiRWalk
database for miRNA-target interactions. Methods Mol Biol.
1182:289–305. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nandan MO, Chanchevalap S, Dalton WB and
Yang VW: Kruppel-like factor 5 promotes mitosis by activating the
cyclin B1/Cdc2 complex during oncogenic Ras-mediated
transformation. FEBS Lett. 579:4757–4762. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu Y, Wen JK, Dong LH, Zheng B and Han M:
Kruppel-like factor (KLF) 5 mediates cyclin D1 expression and cell
proliferation via interaction with c-Jun in Ang II-induced VSMCs.
Acta Pharmacol Sin. 31:10–18. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bao B, Rodriguez-Melendez R and Zempleni
J: Cytosine methylation in miR-153 gene promoters increases the
expression of holocarboxylase synthetase, thereby increasing the
abundance of histone H4 biotinylation marks in HEK-293 human kidney
cells. J Nutr Biochem. 23:635–639. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ghaleb AM, Nandan MO, Chanchevalap S,
Dalton WB, Hisamuddin IM and Yang VW: Kruppel-like factors 4 and 5:
The yin and yang regulators of cellular proliferation. Cell Res.
15:92–96. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dong JT and Chen C: Essential role of KLF5
transcription factor in cell proliferation and differentiation and
its implications for human diseases. Cell Mol Life Sci.
66:2691–2706. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu R, Dong JT and Chen C: Role of KLF5 in
hormonal signaling and breast cancer development. Vitam Horm.
93:213–225. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bialkowska AB, Liu Y, Nandan MO and Yang
VW: A colon cancer-derived mutant of Kruppel-like factor 5 (KLF5)
is resistant to degradation by glycogen synthase kinase 3β (GSK3β)
and the E3 ubiquitin ligase F-box and WD repeat domain-containing
7α (FBW7 α). J Biol Chem. 289:5997–6005. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Frigo DE, Sherk AB, Wittmann BM, Norris
JD, Wang Q, Joseph JD, Toner AP, Brown M and McDonnell DP:
Induction of Kruppel-like factor 5 expression by androgens results
in increased CXCR4-dependent migration of prostate cancer cells in
vitro. Mol Endocrinol. 23:1385–1396. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li X, Liu X, Xu Y, Liu J, Xie M, Ni W and
Chen S: KLF5 promotes hypoxia-induced survival and inhibits
apoptosis in non-small cell lung cancer cells via HIF-1α. Int J
Oncol. 45:1507–1514. 2014.PubMed/NCBI
|
|
23
|
Nakaya T, Ogawa S, Manabe I, Tanaka M,
Sanada M, Sato T, Taketo MM, Nakao K, Clevers H, Fukayama M, et al:
KLF5 regulates the integrity and oncogenicity of intestinal stem
cells. Cancer Res. 74:2882–2891. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhu N, Gu L, Findley HW, Chen C, Dong JT,
Yang L and Zhou M: KLF5 Interacts with p53 in regulating survivin
expression in acute lymphoblastic leukemia. J Biol Chem.
281:14711–14718. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li Q, Dong Z, Zhou F, Cai X, Gao Y and
Wang LW: Kruppel-like factor 5 promotes lung tumorigenesis through
upregulation of Sox4. Cell Physiol Biochem. 33:1–10. 2014.
View Article : Google Scholar : PubMed/NCBI
|