Introduction
Severe bacterial infection may contribute to
systemic dysfunction, including heart failure, which is the
predominant cause of morbidity and mortality in patients with
sepsis. Lipopolysaccharide (LPS) is a major component of the
bacterial outer membrane and has been shown to play an important
role in the inflammatory response in the cardiovascular system
(1). Under septic conditions, the
LPS-induced activation of toll-like receptor 4 (TLR-4) results in
the enhanced production of proinflammatory cytokines and results in
myocardial dysfunction (2). Previous
studies have shown that LPS has adverse effect on cardiomyoblasts,
which induces the activation of TLR-4 and triggers nuclear
factor-κB (NF-κB) signaling and results in increased the expression
of proinflammatory cytokines such as interleukin-1β (IL-1β), IL-6,
and tumor necrosis factor-α (TNF-α) (3–5). During
sepsis, the pathological production of inducible nitric oxide
synthase (iNOS) and cyclooxygenase-2 (COX-2) may also have harmful
effects on cardiomyocyte and impair cardiac contractile function
(6,7). Interventions for sepsis involving
anti-inflammatory agents have been demonstrated to reduce risk of
cardiovascular complications (8,9).
Glucagon-like peptide-1 (GLP-1) and GLP-1 receptors
(GLP-1Rs) serve crucial function in glucose-stimulated insulin
release, leading to great interest in their use for glycemic
control (10). Furthermore, recent
studies have shown that GLP-1 reduces inflammation and oxidative
stress in endothelial cells (11,12). The
importance of GLP-1 in glucose homeostasis is emphasized by the
inhibitors of dipeptidyl peptidase-4 (DPP-4); enzymes which have
been found to exert positive effects on cardioprotection in
previous studies (13,14). DPP-4 inhibitors may improve cardiac
outcomes following myocardial infarction via cytoprotective
pathways (15). Notably, DPP-4
inhibitors have been demonstrated to exert antiatherogenic effects
on improving endothelial function through augmenting GLP-1
activity, and the anti-inflammatory effect of DPP-4 inhibitors has
been suggested in preclinical and clinical studies of type 2
diabetes and coronary artery disease (16,17).
However, the role of DPP-4 inhibitors in septic inflammation, which
may lead to cardiovascular complications, remains unclear.
The aim of the present study was to clarify whether
septic complications may be effectively reduced by DPP-4
therapeutic targeting of LPS-induced immune responses. It is
conceivable that intervention in inflammatory process is beneficial
for sepsis-associated cardiac dysfunctions. We investigated whether
the DPP-4 inhibitor, sitagliptin, was able to exert inhibitory
effects on an LPS-stimulated inflammatory response in
cardiomyoblasts.
Materials and methods
Cell culture
H9c2 cells, a rat ventricular myoblast cell line,
were obtained from the Food Industry Research and Development
Institute (Hsinchu, Taiwan). The cells were maintained in Gibco
Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with 10% Gibco fetal bovine
serum (FBS; Thermo Fisher Scientific, Inc.) in humidified air (5%
CO2) at 37°C. H9c2 cells were incubated overnight prior
to treatment. Following stimulation with 10 µg/ml LPS
(Sigma-Aldrich, St. Louis, MO, USA) for 4 h, cells were treated
with the DPP-4 inhibitor sitagliptin (0, 0.1, 0.5, 1, 2 and 4 µM;
Sigma-Aldrich) and accompanied LPS at 37°C for 20 h.
Cell viability assay
Cell viability was determined using an MTT assay.
Briefly, H9c2 cells were seeded at a density of 1×105
cells/well in 24-well plates 24 h prior to treatments. Following
treatment, the culture medium was replaced with MTT solution
(Sigma-Aldrich), consisting of 5 mg/ml stock solution in PBS,
diluted with DMEM to a final concentration of 0.5 mg/ml. After 3 h
incubation at 37°C the supernatant was aspirated, and the formazan
produced was solubilized in dimethyl sulfoxide (Sigma-Aldrich). The
resulting absorbance was measured at a wavelength of 540 nm with
background subtraction at 650 nm using an EMax® Endpoint
ELISA Microplate Reader (Molecular Devices, LLC, Sunnyvale, CA,
USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Following treatment, total RNA was extracted using
TRIzol reagent (Ambion; Thermo Fisher Scientific, Inc.). Genomic
DNA was removed by adding nanopure water, RNA, DNase buffer and
DNase (Qiagen, Hilden, Germany) to samples. The quantity of each
RNA sample was determined using a Qubit fluorometer (Thermo Fisher
Scientific, Inc.). Reverse transcription was performed in a 20-µl
reaction system containing 200 ng total RNA using high capacity
cDNA reverse transcription kits (cat. no. 4368814; Applied
Biosystems; Thermo Fisher Scientific, Inc.). Relative
quantification of inflammation mediators and apoptosis indicators
were assessed by qPCR, which was performed using SYBR Green mix
Bio-Rad Laboratories, Inc., Hercules, CA, USA), using an ABI 7900HT
Real-Time PCR System (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The primer sequences for each gene were supplied by Gene
Probe Technologies, Inc. (Gaithersburg, MD, USA) and are presented
in Table I. The reaction mixture for
RT-qPCR was as follows: SYBR Green, 10 µl; forward primer, 0.4 µl;
reverse primer, 0.4 µl; cDNA, 2 µl; and nuclease-free
H2O, 7.2 µl. The reaction system for PCR used the
following thermal cycle conditions: 35 Cycles of 94°C for 30 sec,
55°C for 30 sec and 72°C for 45 sec. Negative controls containing
all PCR components without template DNA were used to ensure that
the reagent mix was free of contamination. Cycle threshold (Cq)
values were determined by automated threshold analysis. The average
Cq, ΔCq, ΔΔCq and RQ were calculated by detection software (7500
Fast System SDS Software; version 1.4; Applied Biosystems; Thermo
Fisher Scientific, Inc.). The RQ (ΔΔCq value) was used to compare
gene expression between samples. A housekeeping gene β-actin was
used as an internal control. Primer sequences are indicated in
Table I.
 | Table I.Primers used for reverse
transcription-quantitative polymerase chain reaction analysis. |
Table I.
Primers used for reverse
transcription-quantitative polymerase chain reaction analysis.
| Gene | Forward primer
(5′-3′) | Reverse primer
(3′-5′) |
|---|
| TNF-α |
CACCGGCAAGGATTCCAA |
CACTCAGGCATCGACATTCG |
| IL-6 |
TCTCCAGCAACGAGGAGAAT' |
TGTGATCTGAAACCTGCTGC |
| IL-1β |
GACCTTCCAGGATGAGGACA |
AGGCCACAGGTATTTTGTCG |
| iNOS |
TGCTTACAGGTCTACGTTCAAGACAT |
CGGCCACCAGCTTCTTCA |
| COX2 |
GGATCCCCAAGGCACAAATAT |
TCGCTTCTGATCTGTCTTGAAAAA |
| NF-κB |
TGAGTCCCGCCCCTTCTAA |
TGATGGTCCCCCCAGAGA |
| β-actin |
ATGCCCCGAGGCTCTCTT |
CAACGTCACACTTCATGATGGA |
Enzyme-linked immunosorbent assay
(ELISA)
Highly specific quantitative sandwich ELISA kits
were used to measure the expression of the cytokines IL-6
(KHC0061), IL-1β (KHC0011)and TNF-α (KAC1751) (all purchased from
Thermo Fisher Scientific, Inc.), according to the manufacturer's
instructions. The optical density of each ELISA sample was
determined at 450 nm, with the correction wavelength of 570 nm,
using a microplate reader. Concentrations of cytokine are
calculated on the basis of a standard curve.
Subcellular fractionation
Cells were washed with PBS and incubated with a
lysis buffer (10 mM HEPES, pH7.6; containing 15 mM KCl, 2 mM
MgCl2, 0.1 mM EDTA, 1 mM dithiothreitol, 0.05% v/v
Igepal CA-630 and 1 mM PMSF, 1 mM sodium orthovanadate, 50 mM
sodium fluoride, 10 µg/ml leupeptin and 10 µg/ml aprotinin;
Sigma-Aldrich) for 10 min at 4°C. Resulting lysates were
centrifuged at 2,500 × g for 10 min at 4°C. The supernatant was
collected and centrifuged at 20,000 × g for 15 min at 4°C,
comprising the cytosolic fraction. Pellets were washed with PBS and
suspended in nuclear buffer 5 mM HEPES, pH7.6, 0.1% v/v Igepal
CA-630, 1 M KCl, 0.1 mM EDTA, 1 mM PMSF, 1 mM sodium orthovanadate,
2 mM sodium fluoride, 10 µg/ml leupeptin and 10 µg/ml aprotinin;
Sigma-Aldrich) followed by a centrifugation at 10,000 × g for 15
min at 4°C. The resulting supernatants were collected, comprising
the nuclear fraction.
Western blot analysis
Following treatment, cells were lysed by incubation
with radioimmunoprecipitation assay buffer (Sigma-Aldrich) for 5
min on ice. Samples were centrifuged at 4°C for 15 min at 16,000 ×
g and the supernatants were collected. The protein concentration
was measured using a Qubit® Protein Assay kit (Thermo
Fisher Scientific, Inc.). Next, ~20 µg protein per sample was
loaded in 10–12% SDS-PAGE, then transferred to PVDF (EMD Millipore,
Billerica, MA, USA) using 100 V for 1 h at 4°C. Blocking was
performed with 5% bovine serum albumin in Tris-buffered saline
(Sigma-Aldrich) with Tween 20 (TBST) and subsequently immunoblotted
with the following primary antibodies overnight at 4°C: Rabbit
polyclonal anti-human NF-κB (sc-372; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA), mouse monoclonal anti-human Actin (A5441;
Sigma-Aldrich) and rabbit polyclonal anti-human histone H3
(ab18521; Abcam, Cambridge, UK) and used at a dilution of 1:1,000.
The blots were then washed with TBST and incubated with horseradish
peroxidase-conjugated donkey anti-mouse (ab97030) and donkey
anti-rabbit (ab97064) secondary antibodies at a 1:5,000 dilution
for 30 min at room temperature. The blots were developed using ECL
Western Blotting Detection Reagents (Amersham; GE Healthcare Life
Sciences, Little Chalfont, UK).
Statistical analysis
Statistical analyses were performed using SPSS
version 13.0 for Windows (SPSS, Inc., Chicago, IL, USA). The value
of each treatment group was presented as a mean with the standard
deviation of triplicates. Data were compared using one-way analysis
of variance with post hoc Tukey-Kramer multiple comparisons
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Effect of DPP-4 inhibitor on viability
of H9c2 cells
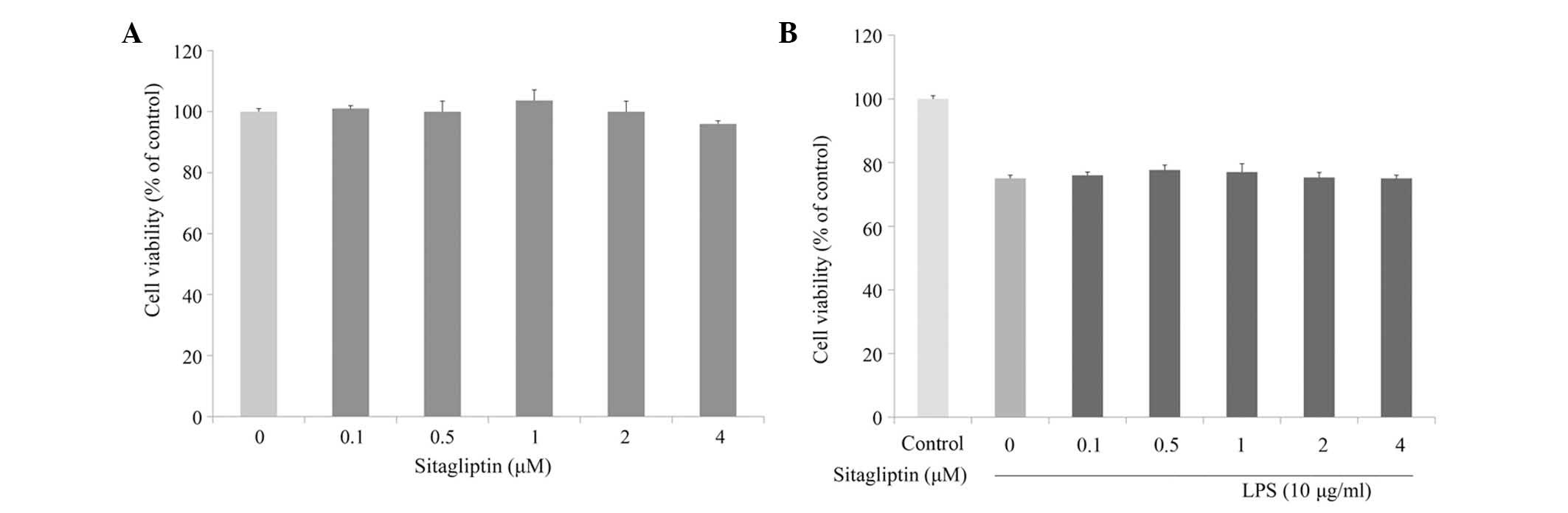
The cytotoxic effect of DPP-4 inhibitor on H9c2 cell
viability was evaluated at various concentrations using MTT assay.
As shown in Fig. 1A, incubation of
H9c2 cells with a serial of concentration of DPP-4 inhibitor (0.1–4
µM) for 24 h slightly affected cell viability. Next, the cytotoxic
effect of DPP-4 inhibitor on LPS-stimulated H9c2 cells was
investigated. Cell viability of the H9c2 cells was slightly
decreased in the presence of LPS; however, DPP-4 inhibitor exerted
no effect on the viability of LPS-treated H9c2 cells (Fig. 1B).
Effect of LPS and the DPP-4 inhibitor
sitagliptin on H9c2 cell morphology
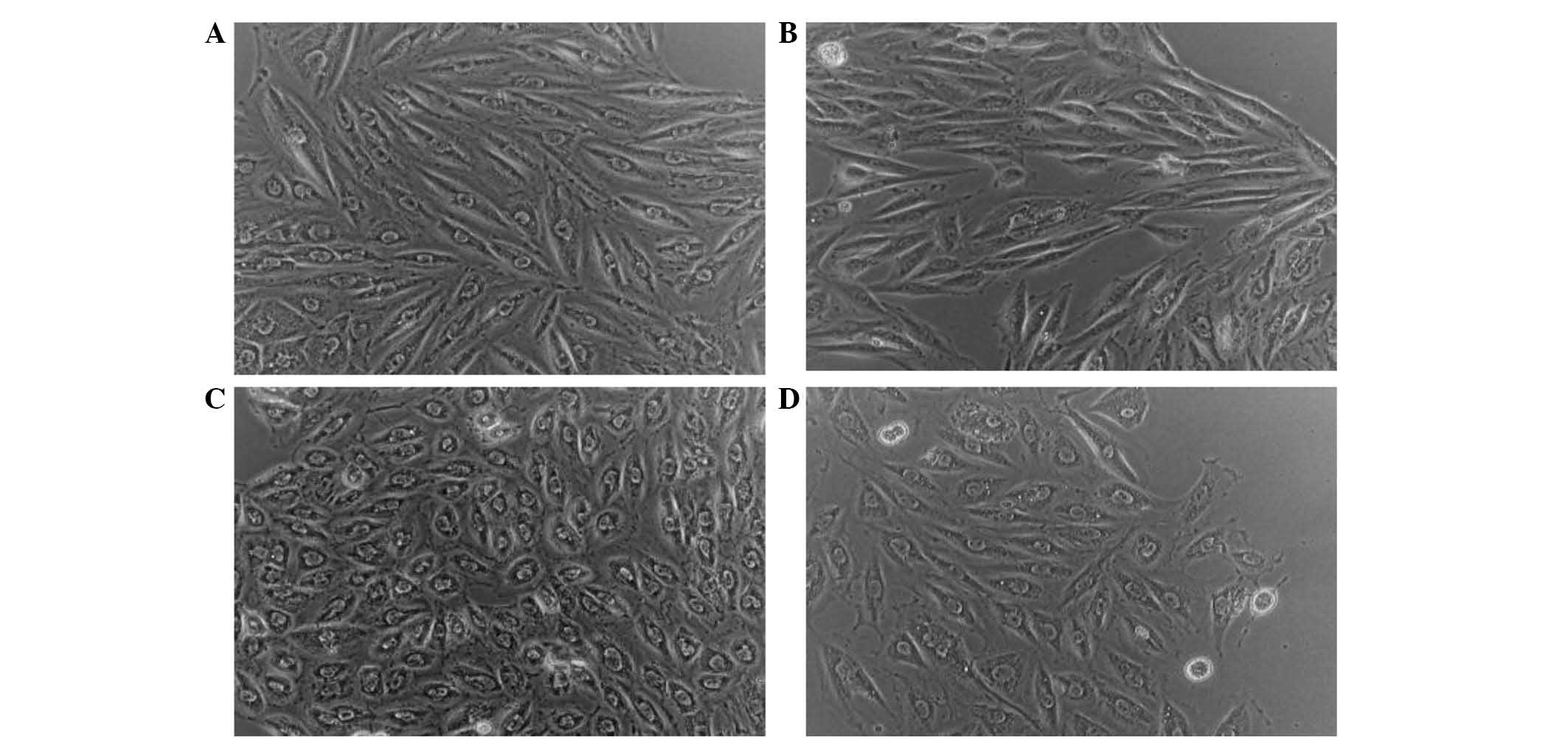
The effect of LPS and sitagliptin on H9c2 cell
morphology were observed. Fig. 2A
shows H9c2 cells without any treatment. When these cells were
treated with sitagliptin alone, there was no apparent change in
cellular shape as shown in Fig. 2B.
However, following LPS stimulation the H9c2 cells exhibited cell
rounding (Fig. 2C), which may
indicate membrane blebbing due to morphological alterations.
However, as a result of the administration of sitagliptin following
LPS stimulation, H9c2 cells exhibited reduced phenotypic responses
(Fig. 2D).
Effect of DPP-4 inhibitor on the
regulation of proinflammatory mediator expression in LPS-treated
H9c2 cells
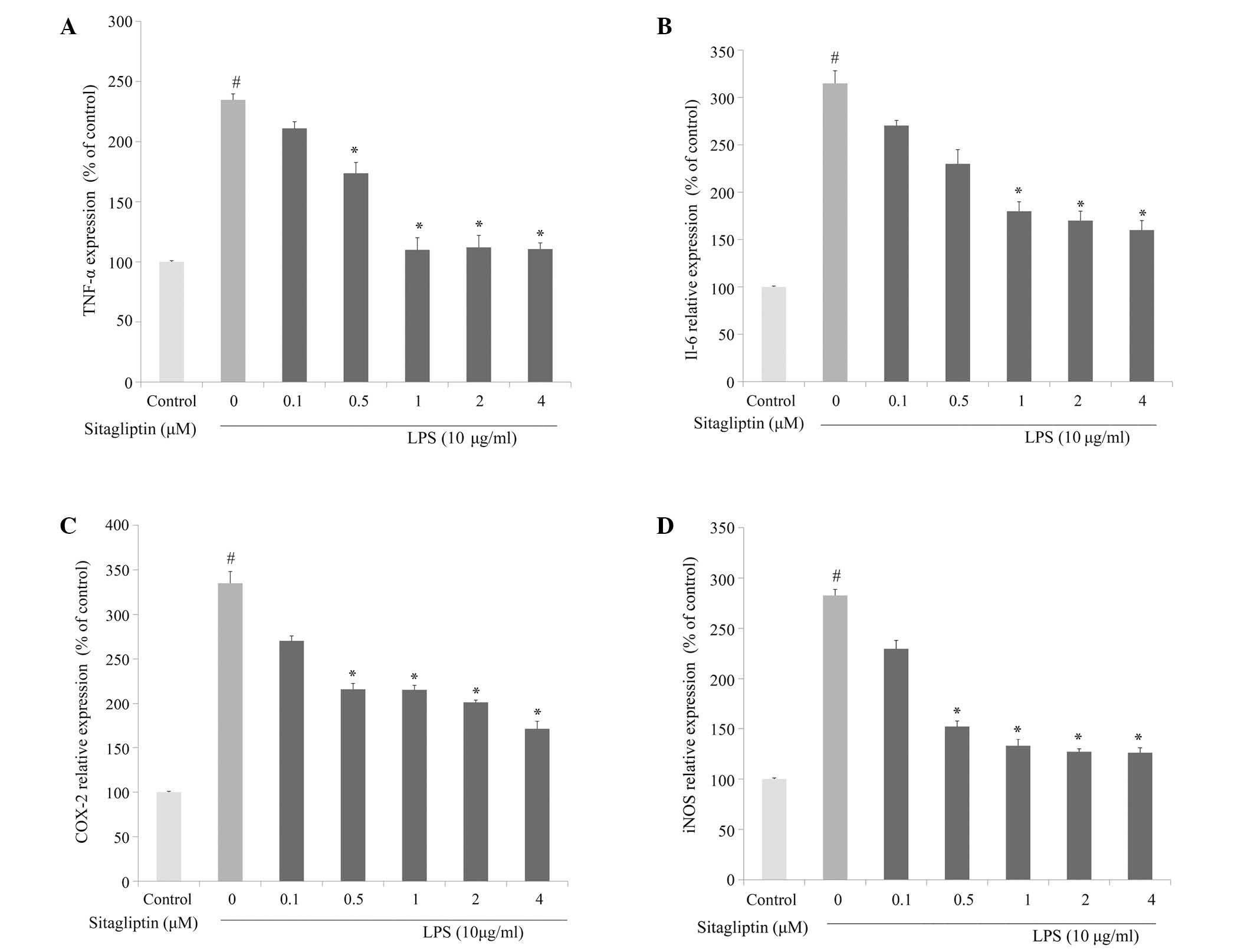
To investigate whether DPP-4 inhibitor alleviates
inflammatory responses in cardiovascular tissue, the changes in the
mRNA expression levels of inflammation-associated genes following
DPP-4 inhibitor treatment in LPS-treated H9c2 cells were evaluated
using qPCR analysis. The elevated mRNA expression of TNF-α was
reduced following treatment with DPP-4 inhibitor (0.1–4 µM)
(Fig. 3A). The mRNA expression of
IL-6 in H9c2 cells was significantly increased in presence of LPS.
The elevation of IL-6 in LPS-treated H9c2 cells was partially
normalized as a result of exposure to DPP-4 inhibitor, and the
alleviation was dose-dependent (Fig.
3B). It is known that LPS induces the activation of COX-2
transcription, leading to a release of prostaglandin E2
(18). The present data showed that
LPS-treated H9c2 cells exhibited a significant increase in mRNA
expression of COX-2. Treatment of LPS-stimulated H9c2 cells with
DPP-4 inhibitor resulted in a suppression of the LPS-elevated
expression of COX-2 (Fig. 3C). The
mRNA expression levels of iNOS in H9c2 were significantly increased
in response to exposure to LPS. The elevated expression of iNOS in
H9c2 was significantly downregulated by DPP-4 inhibitor treatment
at 0.5, 1, 2 and 4 µM (Fig. 3D). The
amelioration of the LPS-induced upregulation of the expression of
TNF-α, IL-6, COX-2 and iNOS by the DPP-4 inhibitor sitagliptin was
dose-dependent.
Effect of DPP-4 inhibitor on the
protein expression of proinflammatory cytokines in LPS-treated H9c2
cells
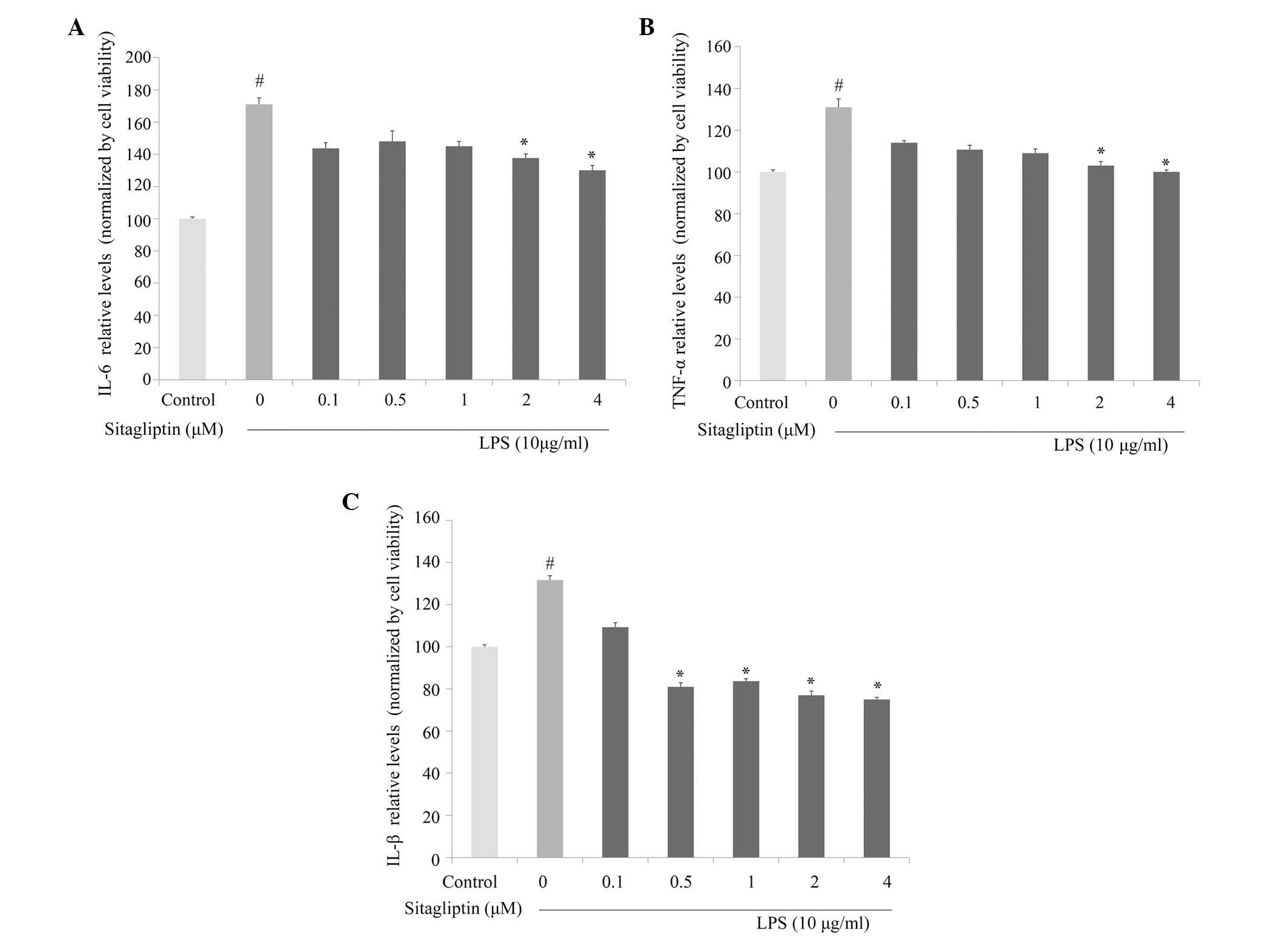
Next, the anti-inflammatory activity of DPP-4
inhibitor against the production of proinflammatory cytokines was
investigated in LPS-treated H9c2 cells. IL-6 and TNF-α production
in culture medium were evaluated using ELISA. As shown in Fig. 4A, IL-6 production was significantly
decreased by DPP-4 inhibitor treatment at concentrations of 2 and 4
µM compared with cells treated with LPS alone. Exposure of H9c2
cells to LPS led to a significant increase in TNF-α secretion,
which is consistent with the observed upregulation of expression of
TNF-α at the mRNA level. Treatment with DPP-4 inhibitor led to
partially normalized protein expression of TNF-α compared with the
cells treated with LPS alone (Fig.
4B). Following stimulation, IL-1β is often upregulated in
association with TNF-α and IL-6 under inflammation conditions. The
LPS-elevated expression of IL-1β in the H9c2 cells was
significantly inhibited by DPP-4 inhibitor treatment at 0.5, 1, 2
and 4 µM (Fig. 4C).
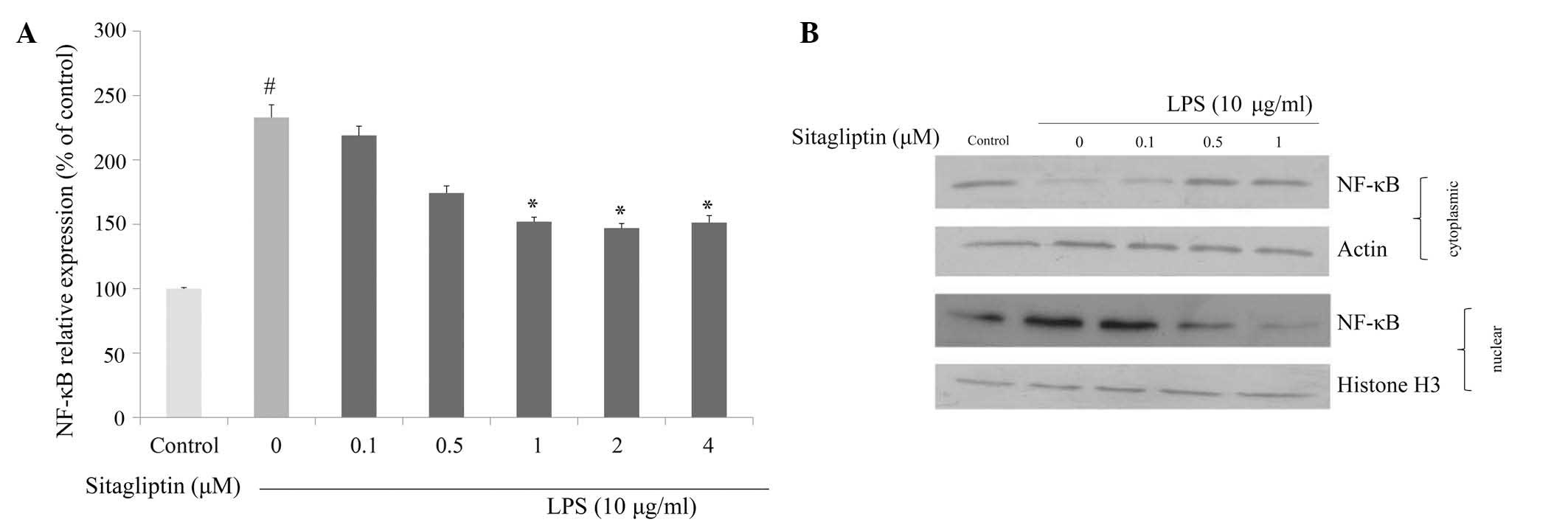
Effect of DPP-4 inhibitor on NF-κB
regulation
It is evident that NF-κB plays a crucial role in
regulating the expression of proinflammatory genes, including TNF-α
and COX-2. We investigated whether DPP-4 inhibitor exerts
anti-inflammatory effect via the inhibition of NF-κB in LPS-treated
H9c2 cells. mRNA expression and nuclear translocation of NF-κB were
examined using qPCR and western blot analyses, respectively. The
mRNA expression of NF-κB in H9c2 cells was significantly
upregulated in the presence of LPS. Exposure of LPS-treated H9c2
cells to DPP-4 inhibitor resulted in suppression of the
LPS-elevated NF-κB expression (Fig.
5A). We also examined the influence of DPP-4 inhibitor on the
translocation of NF-κB in response to LPS exposure. The data from
western blot analysis indicated that LPS triggered nuclear
translocation of NF-κB in H9c2 cells, and that degree of
translocation of NF-κB was reduced by DPP-4 inhibitor treatment
(Fig. 5B).
Discussion
The expression of inflammatory markers is increased
in sepsis patients with cardiovascular dysfunction. Prior studies
have shown elevations in inflammatory marker expression in
infectious individuals who undergo systemic bacterial infection
(19,20). In patients with endoxemia and sepsis,
circulating LPS induces the elevation of proinflammatory cytokines,
which may contribute to myocardial depression (21). It was shown that LPS participates in
the inflammatory response by resulting in enhanced expression of
proinflammatory cytokines such as TNF-α and causes dysfunction in
the cardiovascular system (22).
Under septic conditions, excessive LPS enhances the
expression of proinflammatory cytokines and chemokine cascades via
the activation of TLR-4. Previous studies have shown that
cardiomyoblasts express TLR-4, through which LPS exerts a adverse
effect mediated by NF-κB signaling, resulting in decreased
cardiomyocyte contractility (23,24). It
has been reported that the regulation of NF-κB was involved in the
amelioration of inflammatory responses by DPP-4 inhibitor (25). Several studies also demonstrated that
DPP-4 inhibitor has beneficial effects on cardiovascular system and
gain the ability to improve renal microvasculature (26,27).
However, the role of DPP-4 inhibition in ameliorating
cardiovascular complication and potential anti-inflammatory
properties during sepsis has not been largely investigated.
Therefore, there is a requirement to determine whether DPP-4
inhibitor exerts direct anti-inflammatory effect on cardiomyoblast
during sepsis-induced inflammation.
The aim of the present study was investigate the
effects of sitagliptin on LPS-induced inflammation in
cardiomyoblasts. The results indicate that sitagliptin inhibited
the increased mRNA expression of inflammatory genes in
LPS-stimulated cardiomyoblasts, including TNF-α, IL-6, COX-2 and
iNOS. Furthermore, the activated expression of NF-κB was
downregulated in the presence of sitagliptin. Additionally,
treatment of LPS-stimulated H9c2 cells with sitagliptin resulted in
the inhibition of the elevated protein expression of TNF-α, IL-6
and IL-1β. A previous study has reported that the increased
expression of proinflammatory cytokines is associated with
mortality and may indicate the severity of sepsis (20). In addition, IL-6 was modulated by
IL-1β and TNF-α in cardiomyocytes due to circulating LPS, via NF-κB
signaling pathway (28). The present
results suggest that LPS significantly induced the mRNA expression
of IL-6 and TNF-α, in addition to stimulating NF-κB activation in
H9c2 cells. These inflammatory cytokines were reduced in the
presence of sitagliptin, suggesting that sitagliptin has inhibitory
effects on inflammation in response to proinflammatory agents such
as LPS, which can lead to cardiovascular depression during sepsis.
The present results are consistent with previous studies which have
shown that sitagliptin can lead to reduced levels of
proinflammatory markers, and may potentially contribute to the
inhibition of cardiovascular complications (29,30).
In addition to proinflammatory cytokines, it has
been demonstrated that sepsis results in significant expression of
COX-2 in cardiomyocytes, with subsequent release of prostaglandin
during myocardial inflammation (31). The present data revealed that the
expression of COX-2 was upregulated by LPS in cardiomyoblasts, and
that this upregulation could be suppressed by sitagliptin. This
result suggested that treatment with sitagliptin inhibits
LPS-induced overexpression of COX-2, leading to a reduction of
prostaglandin release. It is evident that endotoxemia is associated
with increased production of NO, mediated by iNOS (32). The increased NO production may reduce
the endothelium-dependent vasodilatory response, due to the
downregulation of endothelial NOS (eNOS) (33). The present results show that the mRNA
expression of iNOS is increased in LPS-treated H9c2 cells, and the
elevated expression of iNOS is inhibited in the presence of
sitagliptin.
Prior studies have shown that the increased
expression of TNF-α in cardiomyocytes during sepsis via the NF-κB
signaling pathway (34). Over
production of TNF-α and LPS stimulation may induce the activity of
NF-κB and exert harmful effects on myocardial cells (35). The current results revealed that
sitagliptin is able to reduce LPS-induced TNF-α production and
inhibit the nuclear translocation of NF-κB. This study demonstrates
that the DPP-4 inhibitor sitagliptin reduces the LPS-stimulated
expression of inflammatory cytokines due, and these results
indicate that DPP-4 has the potential to serve as a target for
reverse cardiac remodeling due to endotoxemia and sepsis.
In conclusion, the present results suggest that the
DPP-4 inhibitor sitagliptin reduced the LPS-induced inflammatory
response, which was mediated by the NF-κB pathway signaling
pathway. Treatment with sitagliptin decreased the protein
expression levels of the inflammatory cytokines TNF-α, IL-6 and
IL-1β, and the mRNA expression of COX-2 and iNOS in
cardiomyoblasts. These findings suggest that DPP-4 inhibitors may
be beneficial to the suppression of septic inflammation, which may
lead to further cardiovascular complications. Further efforts to
determine the most effective application of DPP-4 inhibitor to
attenuate LPS-induced inflammatory response are required, and this
strategy may provide novel therapies for treating septic patients
and reducing subsequent cardiomyopathy.
Acknowledgements
The present study was supported by research grants
(grant no. 101XDAA00012) from Taipei City Hospital.
References
|
1
|
Jirik FR, Podor TJ, Hirano T, Kishimoto T,
Loskutoff DJ, Carson DA and Lotz M: Bacterial lipopolysaccharide
and inflammatory mediators augment IL-6 secretion by human
endothelial cells. J Immunol. 142:144–147. 1989.PubMed/NCBI
|
|
2
|
Fallach R, Shainberg A, Avlas O, Fainblut
M, Chepurko Y, Porat E and Hochhauser E: Cardiomyocyte toll-like
receptor 4 is involvedin heart dysfunction following septic shock
or myocardial ischemia. J Mol Cell Cardiol. 48:1236–1244. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lu YC, Yeh WC and Ohashi PS: LPS/TLR4
signal transduction pathway. Cytokine. 42:145–151. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Du J, An J, Wei N, Guan T, Pritchard KA Jr
and Shi Y: Increased resistance to LPS-induced myocardial
dysfunction in the Brown Norway rats versus Dahl S rats: Roles of
inflammatory cytokines and nuclear factor kappaB pathway. Shock.
33:332–336. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Knuefermann P, Nemoto S, Baumgarten G,
Misra A, Sivasubramanian N, Carabello BA and Vallejo JG: Cardiac
inflammation and innate immunity in septic shock: Is there a role
for toll-like receptors? Chest. 121:1329–1336. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Panaro MA, Acquafredda A, Cavallo P,
Cianciulli A, Saponaro C and Mitolo V: Inflammatory responses in
embryonal cardiomyocytesexposed to LPS challenge: An in vitro model
of deciphering the effects of LPS on the heart. Curr Pharm Des.
16:754–765. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Grandel U, Hopf M, Buerke M, Hattar K,
Heep M, Fink L, Bohle RM, Morath S, Hartung T, Pullamsetti S, et
al: Mechanisms of cardiac depression caused by lipoteichoic acids
from staphylococcus aureus in isolated rat hearts. Circulation.
112:691–698. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Reddy AB, Srivastava SK and Ramana KV:
Anti-inflammatory effect of aldose reductase inhibition in murine
polymicrobial sepsis. Cytokine. 48:170–176. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
von Rosenstiel N, von Rosenstiel I and
Adam D: Management of sepsis and septic shock in infants and
children. Paediatr Drugs. 3:9–27. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cariou B: Pleiotropic effects of insulin
and GLP-1 receptor agonists: Potential benefits of the association.
Diabetes Metab. 41:6S28–6S35. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ceriello A, Novials A, Ortega E, Canivell
S, La Sala L, Pujadas G, Esposito K, Giugliano D and Genovese S:
Glucagon-like peptide 1 reduces endothelial dysfunction,
inflammation and oxidative stress induced by both hyperglycemia and
hypoglycemia in type 1 diabetes. Diabetes Care. 36:2346–2350. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shiraki A, Oyama J, Komoda H, Asaka M,
Komatsu A, Sakuma M, Kodama K, Sakamoto Y, Kotooka N, Hirase T and
Node K: The glucagon-like peptide 1 analog liraglutide reduces
TNF-α-induced oxidative stress and inflammation in endothelial
cells. Atherosclerosis. 221:375–382. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee TI, Kao YH, Chen YC, Huang JH, Hsu MI
and Chen YJ: The dipeptidyl peptidase-4 inhibitor-sitagliptin
modulates calcium dysregulation, inflammation and PPARs in
hypertensive cardiomyocytes. Int J Cardiol. 168:5390–5395. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chinda K, Palee S, Surinkaew S,
Phornphutkul M, Chattipakorn S and Chattipakorn N: Cardioprotective
effect of dipeptidyl peptidase-4 inhibitor during
ischemia-reperfusion injury. Int J Cardiol. 167:451–457. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang XM, Yang YJ and Wu YJ: The emerging
role of dipeptidyl peptidase-4 inhibitors in cardiovascular
protection: Current position and perspectives. Cardiovasc Drugs
Ther. 27:297–307. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu L, Liu J, Wong WT, Tian XY, Lau CW,
Wang YX, Xu G, Pu Y, Zhu Z, Xu A, et al: Dipeptidyl peptidase 4
inhibitor sitagliptin protects endothelial function in hypertension
through a glucagon-like peptide 1-dependent mechanism.
Hypertension. 60:833–841. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Matsubara J, Sugiyama S, Akiyama E,
Iwashita S, Kurokawa H, Ohba K, Maeda H, Fujisue K, Yamamoto E,
Kaikita K, et al: Dipeptidyl peptidase-4inhibitor, sitagliptin,
improves endothelial dysfunction in association with its
anti-inflammatory effects in patients with coronary artery disease
and uncontrolled diabetes. Circ J. 77:1337–1344. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Panaro MA, Pricci M, Meziani F, Ragot T,
Andriantsitohaina R, Mitolo V and Tesse A: Cyclooxygenase-2-derived
prostacyclin protective role on endotoxin-induced mouse
cardiomyocyte mortality. Cardiovasc Toxicol. 11:347–356. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lobo SM and Lobo FR: Markers and mediators
of inflammatory response in infection and sepsis. Rev Bras Ter
Intensiva. 19:210–215. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ashare A, Powers LS, Butler NS, Doerschug
KC, Monick MM and Hunninghake GW: Anti-inflammatory response is
associated with mortality and severity of infection in sepsis. Am J
Physiol Lung Cell Mol Physiol. 288:L633–L640. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tavener SA and Kubes P: Cellular and
molecular mechanisms underlying LPS-associated myocyte impairment.
Am J Physiol Heart Circ Physiol. 290:H800–H806. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jatta K, Wågsäter D, Norgren L, Stenberg B
and Sirsjö A: Lipopolysaccharide-induced cytokine and chemokine
expression in human carotid lesions. J Vasc Res. 42:266–271. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nemoto S, Vallejo JG, Knuefermann P, Misra
A, Defreitas G, Carabello BA and Mann DL: Escherichia coli
LPS-induced LV dysfunction: Role of toll-like receptor-4 in the
adult heart. Am J Physiol Heart Circ Physiol. 282:H2316–H2323.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Panaro MA, Gagliardi N, Saponaro C,
Calvello R, Mitolo V and Cianciulli A: Toll-like receptor 4
mediates LPS-induced release of nitric oxide and tumor necrosis
factor-alpha by embryonal cardiomyocytes: Biological significance
and clinical implications in human pathology. Curr Pharm Des.
16:766–774. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kodera R, Shikata K, Takatsuka T, Oda K,
Miyamoto S, Kajitani N, Hirota D, Ono T, Usui HK and Makino H:
Dipeptidyl peptidase-4 inhibitor ameliorates early renal injury
through its anti-inflammatory action in a rat model of type 1
diabetes. Biochem Biophys Res Commun. 443:828–833. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
DeNicola M, Du J, Wang Z, Yano N, Zhang L,
Wang Y, Qin G, Zhuang S and Zhao TC: Stimulation of glucagon-like
peptide-1 receptor through exendin-4 preserves myocardial
performance and prevents cardiac remodeling in infarcted
myocardium. Am J Physiol Endocrinol Metab. 307:E630–E643. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang WJ, Chang CH, Sun MF, Hsu SF and Weng
CS: DPP-4 inhibitor attenuates toxic effects of indoxyl sulfate on
kidney tubular cells. PLoS One. 9:e934472014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vona-Davis L, Zhu X, Yu AK and McFadden
DW: Modulation of interleukin-6 in cardiac myoblasts during
endotoxemia. J Surg Res. 112:91–96. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Makdissi A, Ghanim H, Vora M, Green K,
Abuaysheh S, Chaudhuri A, Dhindsa S and Dandona P: Sitagliptin
exerts an antinflammatory action. J Clin Endocrinol Metab.
97:3333–3341. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zeng Y, Li C, Guan M, Zheng Z, Li J, Xu W,
Wang L, He F and Xue Y: The DPP-4 inhibitor sitagliptin attenuates
the progress of atherosclerosis in apolipoprotein-E-knockout mice
via AMPK-and MAPK-dependent mechanisms. Cardiovasc Diabetol.
13:322014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Frazier WJ, Xue J, Luce WA and Liu Y: MAPK
signaling drives inflammation in LPS-stimulated cardiomyocytes: The
route of crosstalk to G-protein-coupled receptors. PLoS One.
7:e500712012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Baumgarten G, Knuefermann P, Schuhmacher
G, Vervölgyi V, von Rappard J, Dreiner U, Fink K, Djoufack C, Hoeft
A, Grohé C, et al: Toll-like receptor 4, nitric oxide, and
myocardial depression in endotoxemia. Shock. 25:43–49. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lee CC, Lin NT, Hsu YH and Chen HI:
Inducible nitric oxide synthase inhibition potentiates multiple
organ dysfunction induced by endotoxin in consciousrats. J
Cardiovasc Pharmacol. 45:396–403. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhu H, Shan L, Schiller PW, Mai A and Peng
T: Histone deacetylase-3 activation promotes tumor necrosis
factor-alpha (TNF-alpha) expression in cardiomyocytes during
lipopolysaccharide stimulation. J Biol Chem. 285:9429–9436. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wright G, Singh IS, Hasday JD, Farrance
IK, Hall G, Cross AS and Rogers TB: Endotoxin stress-response in
cardiomyocytes: NF-kappaB activation and tumor necrosis
factor-alpha expression. Am J Physiol Heart Circ Physiol.
282:H872–H879. 2002. View Article : Google Scholar : PubMed/NCBI
|