Introduction
Acute myeloid leukemia (AML) remains the most common
acute leukemia among adults (1). It
is a heterogeneous hematological malignancy characterized by the
clonal expansion of myeloid blasts in peripheral blood, bone marrow
(BM), and/or other tissues. Despite the high response rate of AML
to chemotherapy (the complete remission rate is ~60–80%) (2), the majority of patients with AML
relapse, which has been attributed to residual disease in the BM
(3). The establishment of separate
regimens for elderly patients, relapsed/refractory patients or
secondary AML patients (who are receiving cytotoxic therapy for
solid tumors or hematologic malignancies) may attenuate the poor
outcomes that are exhibited by these groups when treated with the
standard cytarabine and an anthracycline. These poor outcomes may
be due to drug resistance, comorbidity, Eastern Cooperative
Oncology Group performance status or treatment-associated mortality
(4,5). The cytarabine, aclarubicin and
granulocyte colony-stimulating factor (G-CSF) (CAG) treatment
regimen was initially applied to these patients groups in Japan
(6), and widespread popularization
in Asia followed (7). Although the
CAG regimen has demonstrated marked therapeutic efficacy (6), the definite mechanisms underlying the
CAG protocol remain poorly understood.
Leukemia cells exhibit an enhanced proliferation
rate and defective apoptosis, as compared with normal cells;
however, they have been shown to undergo spontaneous apoptosis when
cultured in vitro, thus suggesting that the microenvironment
may have a significant protective role (8). Leukemia cells reside in BM niches,
which are comprised of endothelial cells, perivascular reticular
cells, osteoblasts, osteoclasts and stromal cells (9,10);
therefore, they are predominantly protected against spontaneous or
drug-induced apoptosis (11).
Interaction between leukemia cells and the BM microenvironment has
been proposed as a potential mechanism for chemotherapy resistance,
which is typically dependent on cell-to-cell contact and soluble
signals, including cytokines, chemokines, growth factors and
adhesion molecules (12,13).
Chemokine stromal-derived factor-1α (SDF-1α), which
is also known as C-X-C chemokine ligand 12, and its cognate
receptor, C-X-C chemokine receptor type 4 (CXCR4), which is also
known as cluster of differentiation (CD)184, are considered to be
critical mediators of BM microenvironment/leukemic cell
interactions (14–16). Binding of SDF-1α induces
conformational changes in CXCR4, resulting in its incorporation
into lipid rafts and subsequent phosphorylation (17). Phosphorylation of CXCR4 activates
cell signaling, which ultimately leads to alterations in gene
transcription with consequent changes in the migration, adhesion,
proliferation, survival and drug resistance of leukemia cells.
There are two strategies for the modulation of CXCR4 expression.
The first relies using a specific CXCR4 antagonist, such as
Plerixafor (AMD3100), to compete with SDF-1α for binding to CXCR4
(18,19); and the second involves blocking CXCR4
expression at the mRNA level using small interfering microRNAs
(miRs). miRs are small, non-coding RNAs (19–25 nucleotides) which
modulate gene expression by targeting mRNA in a sequence-specific
manner, leading to mRNA degradation or translational repression
(20). Previous studies have
suggested that miR-146a may have a pivotal role in downregulating
CXCR4 expression (21,22).
G-CSF, which is the main growth factor for the
regulation of the proliferation and differentiation of myeloid
cells, is a common agent used for mobilizing neutrophils, immature
myeloid cells and hematopoietic stem cells (HSCs) from the BM into
the peripheral blood. Previous studies have verified the role of
G-CSF in CXCR4 downregulation (23,24), and
alterations in the expression levels of specific miRs, including
miR-146a, have been reported during G-CSF-mediated mobilization of
HSCs (25). The present study aimed
to investigate the ability of G-CSF to inhibit CXCR4/SDF-1α
signaling, as well as the underlying molecular mechanisms, in order
to overcome stromal cell-mediated drug resistance in AML.
Materials and methods
Cell culture
The HL-60 human acute promyelocytic leukemia cell
line was a gift from the Institute of Hematology at the China
Academy of Chinese Medical Sciences (Beijing, China). HL-60 cells
were cultured in Iscove's modified Dulbecco's medium (IMDM; Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with
20% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.)
at 37°C in a humidified incubator with 5% CO2. The HS-5
human BM/stromal cell line was purchased from ATCC (Manassas, VA,
USA). HS-5 cells were cultured in Dulbecco's modified Eagle's
medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.) supplemented
with 10% FBS in a humidified incubator at 37°C with 5%
CO2.
Reagents and antibodies
SDF-1α, AMD3100, recombinant human G-CSF, bovine
serum albumin (BSA) and cytarabine were purchased from
Sigma-Aldrich (St. Louis, MO, USA). Phycoerythrin (PE)-conjugated
mouse anti-human CD184 (clone 12G5; 561733), peridinin chlorophyll
protein complex (PerCP)-conjugated anti-CD45 (clone 2D1; 561047)
and PE-conjugated IgG2a isotypic control (555574) monoclonal
antibodies (mAb) were purchased from BD Pharmingen (San Diego, CA,
USA). Rabbit anti-human CXCR4 mAb (sc-9046) and horseradish
peroxidase (HRP)-conjugated goat anti-rabbit immunoglobulin G
(sc-2004) were purchased from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). Mouse anti-human β-tubulin mAb (KM9001T) was
purchased from Sungene Biotech Co., Ltd. (Tianjin, China). β-actin
mAB (P3002) was purchased from Abmart (Arlington, MA, USA).
Treatment of HL-60 cells
For functional assays, HL-60 cells were divided into
four groups, as follows: i) Control group (medium alone); ii) G-CSF
group (50 ng/ml G-CSF for 48 h); iii) AMD3100 group (5 µg/ml
AMD3100 for 30 min); and iv) G-CSF plus AMD3100 group (initially
cultured with 50 ng/ml G-CSF for 48 h, then with 5 µg/ml AMD3100
for 30 min). The group treated with AMD3100 only was used as a
positive control.
Migration assay
Migration assays were performed using 6.5 mm
Transwell assays with 8.0 µm pore polycarbonate membrane inserts
(Sigma-Aldrich). In order to increase the sensitivity of the cells
to the SDF1-α chemoattractant and reduce non-specific migration
caused by serum, HL-60 cells were starved for 24–48 h prior to
performing the migration assays, and the FBS was replaced with 0.5%
BSA. Briefly, the lower chamber was supplemented with 600 µl IMDM
medium containing 100 ng/ml SDF-1α or 600 µl HS-5 supernatant.
Assay medium (IMDM with 0.5% BSA) without SDF-1α was used as a
baseline control. Subsequently, 5×105 cells in 100 µl
medium from all groups were added to the upper chamber and
incubated for 3 h under 37°C and 5% CO2 culture
conditions. Following this, cells that had transmigrated into the
lower chamber were collected and counted by flow cytometry (BD
Pharmingen) at high flow for 20 sec. Control cells
(5×105 in 600 µl medium) were added to the lower chamber
and counted under the same conditions. Results are shown as
migration index [mean ± standard error of the mean (SEM)], which
represents the ratio of the number of cells that transmigrated in
the presence of chemoattractant and the number of cells that
transmigrated in the absence of chemoattractant.
Adhesion assay
Firstly, a stromal cell layer composed of HS-5 cells
was established by adding 1×105 cells/well to a 12-well
plate and culturing for 48 h until 80–90% confluence. Subsequently,
HL-60 cells (1×105 cells/well) in the different groups
were added to the HS-5 cell layer. Following co-culturing for 3 h,
non-adherent cells in the suspension were removed. Wells were
washed three times with DMEM and adherent cells were detached by
the addition of 0.5 ml 0.25% trypsin-ethylenediaminetetraacetic
acid (EDTA; Gibco; Thermo Fisher Scientific, Inc.) to each well.
Subsequently, the suspension of the mixed cells group (HL60 and
HS-5) was centrifuged at 1,400 × g at 4°C for 10 min and the
supernatant was discarded. Cells in the pellet sediment were
resuspended in 100 µl DMEM and stained with 4 µl PerCP-conjugated
anti-CD45 mAb prior to counting by flow cytometry at high flow for
20 sec. An aliquot of the untreated cell population was counted
under similar conditions as a control. Adhesion was calculated as a
percentage of the number of initial cells (mean ± SEM).
HS-5/HL-60 co-culture models and drug
treatment
Firstly, a HS-5/HL-60 direct-contact co-culture
model was established. Briefly, HS-5 cells (1×105
cells/well) were seeded onto 12-well plates two days prior to
experiments and incubated at 37°C with 5% CO2. Following
confirmation of the confluence of the stromal layer by phase
contrast microscopy (CX23; Olympus Corporation, Tokyo, Japan),
HL-60 cells (1×105 cells/well) from the various groups
were added to HS-5 layers in the presence or absence of 0.05 µM
cytarabine. Following 48-h incubation, the cells were detached
using 0.25% trypsin-EDTA, washed twice with phosphate-buffered
saline (PBS) and resuspended in RPMI 1640 medium (Gibco; Thermo
Fisher Scientific, Inc.) at a density of 5×105 cells/ml,
since the manufacturer's protocol outlines that IMDM has been shown
to influence optical density (OD) results.
Subsequently, the viability and apoptosis of the
cells was assessed. For the viability assay, HS-5 cells
(1×105 cells/well) were cultured in the absence of HL-60
cells as baseline control. For the apoptosis assay, the cells were
stained with PerCP-conjugated anti-CD45 mAb to separate HL-60 cells
from HS-5 cells.
The present study also established a HS-5/HL-60
indirect-contact co-culture model. Briefly, HL-60 cells
(1×105cells/well) from the various groups were cultured
with HS-5 supernatant or medium alone in the presence or absence of
0.05 µM cytarabine for comparison. Following 48-h incubation, the
cells were resuspended in RPMI 1640 medium at a density of
5×105 cells/ml, and viability and apoptosis assays were
performed.
Viability and apoptosis assays
Cell viability was assessed using a Cell Counting
kit-8 (CCK-8) assay (Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan). Briefly, 100 µl HL-60 cell suspension
(5×105 cells/ml) per well was seeded into a 96-well
plate and 10 µl CCK-8 solution was added to each well, followed by
incubation under culture conditions for 4 h. The absorbance of each
well was determined at OD 490 and 630 nm using a Multiskan
microplate reader (51119100; Thermo Fisher Scientific, Inc.).
Results were expressed as percentages of control values.
Apoptosis was assessed by flow cytometry using a
Alexa Fluor® 488 Annexin V Apoptosis kit (Invitrogen;
Thermo Fisher Scientific, Inc.), according to the manufacturer's
protocol. Briefly, HL-60 cells were washed with cold PBS and
resuspended in 1X Annexin V-binding buffer at a density of
5×105 cells/ml. Subsequently, 5.0 µl Alexa
Fluor® 488 Annexin V was added to 100 µl cell
suspensions, after which the samples were incubated at room
temperature for 15 min, followed by the addition of 400 µl 1X
Annexin-binding buffer and gentle mixing. Samples were maintained
on ice prior to analysis by flow cytometry. Percentage of apoptotic
cells was calculated using the following formula: Apoptotic cells
(%) = number of Annexin V+ HL-60 cells/total number of
input cells × 100.
Surface expression of CXCR4 on HL-60
cells
In order to evaluate CXCR4 surface expression, HL-60
cells (1×106 cells in 100 µl medium) from the various
groups were washed with PBS and labeled using 1.25 µl PE-conjugated
anti-CXCR4. Subsequently, the cells were analyzed by flow cytometry
using the FACSCalibur™ system (343020; BD Pharmingen) and Cell
Quest 5.1 software (BD Pharmingen) for data acquisition and
analysis. CXCR4 expression was assessed by comparison with cells
incubated with 1.25 µl PE-conjugated IgG2a isotypic control
antibody. Results are presented as the mean fluorescence intensity,
which represents the ratio between the mean fluorescence values
observed for the CXCR4-labeled cells and the cells labeled with the
isotypic control.
Western blotting
HL-60 cells were lysed using
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology, Haimen, China) containing 50 mM Tris (pH 7.4), 0.1%
sodium dodecyl sulfate (SDS) and 1 mM phenylmethylsulfonyl fluoride
protease inhibitor. Lysates were maintained on ice for 30 min
following centrifugation at 14,000 × g for 3 min at 4°C. Protein
concentration of the supernatant was determined using a
Bicinchoninic Acid Protein Assay kit (Beyotime Institute of
Biotechnology), according to the manufacturer's protocol. Total
protein (30 µg) from each sample was separated by 10%
SDS-polyacrylamide gel electrophoresis and transferred to
polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA,
USA). Membranes were blocked with 5% non-fat milk in Tris-buffered
saline containing Tween-20 (TBST; Sigma-Aldrich) at room
temperature for 30 min, after which the membranes were incubated
overnight at 4°C with anti-CXCR4 (1:250) and β-tubulin (1:3,000)
primary antibodies. Following rinsing three times with 20 ml TBST
for 5 min, the membranes were incubated with HRP-conjugated goat
anti-rabbit immunoglobulin G secondary antibody (1:1,500) for 1 h
at room temperature. Bands were detected by Western lightning
enhanced chemiluminescence reagent (PerkinElmer, Inc., Waltham, MA,
USA). Results were analyzed on a Tanon 5500 Chemiluminescence
Imaging system (Tanon Science and Technology Co., Ltd., Shanghai,
China) using ImageCal 4.0 software (Tanon Science and Technology
Co., Ltd.) and were normalized to β-tubulin (1:3,000).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from HL-60 cells using
TRIzol reagent (Takara Biotechnology Co., Ltd., Dalian, China),
according to the manufacturer's protocols. After DNase I treatment
(Thermo Fisher Scientific, Inc.), cDNA was synthesized using
RevertAid Reverse Transcriptase (Thermo Fisher Scientific, Inc.).
PCR was performed using SYBR Green qPCR Mix (Dongsheng Biotech Co.,
Ltd., Guangzhou, China) on a Bio-Rad iQ5 PCR thermal cycler
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). Primers are shown
in Table I, with the exception of
the primer sequences for miR-146a, which were unavailable. qPCR was
performed using a reaction volume of 20 µl containing 10 µl SYBR
Green (2X), 1 µl 0.5 µg/µl primers, 1 µl cDNA template and 8 µl
water. PCR conditions were as follows: Denaturation at 95°C for 2
min, followed by 40 cycles of denaturation at 95°C for 15 sec,
annealing at 60°C for 20 sec and extension at 72°C for 20 sec, with
final elongation at 72°C for 5 min. Each gene was tested and the
respective melting curves were detected. All samples were measured
in triplicate. Expression levels of CXCR4 and miR-146a were
normalized to human β-actin and RNU6A reference gene levels,
respectively. Mean Cq values from each sample were normalized
against the mean Cq value of the reference genes. Relative
expression levels were acquired using the 2−ΔΔCq method
(26).
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| Gene | Forward
(5′-3′) | Reverse
(5′-3′) | Product size
(bp) |
|---|
| h-ACTB |
GGCACTCTTCCAGCCTTCC |
GAGCCGCCGATCCACAC | 255 |
| CXCR4 |
TCATCAGTCTGGACCGCTACC |
GACGCCAACATAGACCACCTT | 95 |
Statistical analysis
Data are presented as the mean ± SEM of at least
three independent experiments. Comparisons were performed using a
two-tailed, unpaired Student's t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
G-CSF reduces the migration of HL-60
cells in response to SDF-1α or HS-5 supernatant
Initially, the potential effect of G-CSF on the
SDF-1α-induced migration of HL-60 cells was examined in
vitro. AMD3100-only treated HL-60 cells were used as a positive
control and assay medium-only treated cells were used as the
baseline. The migration index was markedly increased upon exposure
of the control HL-60 cells to medium containing 100 ng/ml SDF-1α,
as compared with HL-60 cells exposed to assay medium without SDF-1α
(1.75±0.08 vs. 1.00±0.02; P<0.01). However, G-CSF and AMD3100
were demonstrated to significantly reduce the SDF-1α-induced
migration of HL-60 cells, as compared with the control (1.27±0.01
and 0.86±0.06 vs. 1.75±0.08, respectively; P<0.01; Fig. 1A). In addition, combination treatment
with G-CSF and AMD3100 reduced the migration index to the greatest
extent, as compared with either agent alone (0.58±0.06 vs.
1.27±0.01 or 0.86±0.06; P<0.01; Fig.
1A). These results suggested that G-CSF may reduce functional
CXCR4 expression and specific responses to SDF-1α in HL-60 cells,
which is similar to AMD3100 treatment.
The ability of HS-5 cells to secrete SDF1-α and
attract HL-60 cells was evaluated using the HS-5 supernatant,
rather than SDF-1α-containing medium, as a chemoattractant. Similar
results were observed as when the SDF1-α medium was used as the
chemoattractant. The migration index of the control HL-60 cells
exposed to HS-5 supernatant was markedly increased, as compared
with the cells exposed to medium alone (3.04±0.05 vs. 1.00±0.04;
P<0.01; Fig. 1B). Conversely,
G-CSF and AMD3100 were demonstrated to significantly reduce the
migration index of HL-60 cells, as compared with the control
(1.75±0.03 and 1.58±0.05 vs. 3.04±0.05, respectively; P<0.01;
Fig. 1B). In addition, combination
treatment with G-CSF and AMD3100 reduced the migration index to the
greatest extent, as compared with either agent alone (0.73±0.03 vs.
1.58±0.05 or 1.75±0.03; P<0.01; Fig.
1B). These data indicated that HS-5 cells may produce SDF-1α to
attract HL-60 cells that express CXCR4 on their surface; whereas
G-CSF may reduce surface CXCR4 expression levels synergistically
with AMD3100.
G-CSF reduces the adhesion of HL-60
cells to HS-5 cells
Since drug resistance is predominantly dependent on
cell-to-cell contact, the present study investigated the potential
effect of G-CSF and/or AMD3100 treatment on the adhesion of HL-60
cells to HS-5 cells. The percentage of HL-60 cells that
specifically adhered to the HS-5 cell layer was significantly
reduced in the G-CSF (10.25±0.03), AMD3100 (11.17±0.35) and G-CSF
plus AMD3100 (8.39±0.03) groups, as compared with the control group
(14.1±0.06; P<0.01; Fig. 2).
Furthermore, the greatest inhibition of adhesion was detected in
the G-CSF plus AMD3100 group (8.39±0.03), as compared with the
G-CSF (10.25±0.03) and AMD3100 (11.17±0.35) groups (P<0.05;
Fig. 2). These results suggest that
HL-60 cells are attracted and adhere to HS-5 cells, and that G-CSF
and AMD3100 are able to block these effects independently and
synergistically.
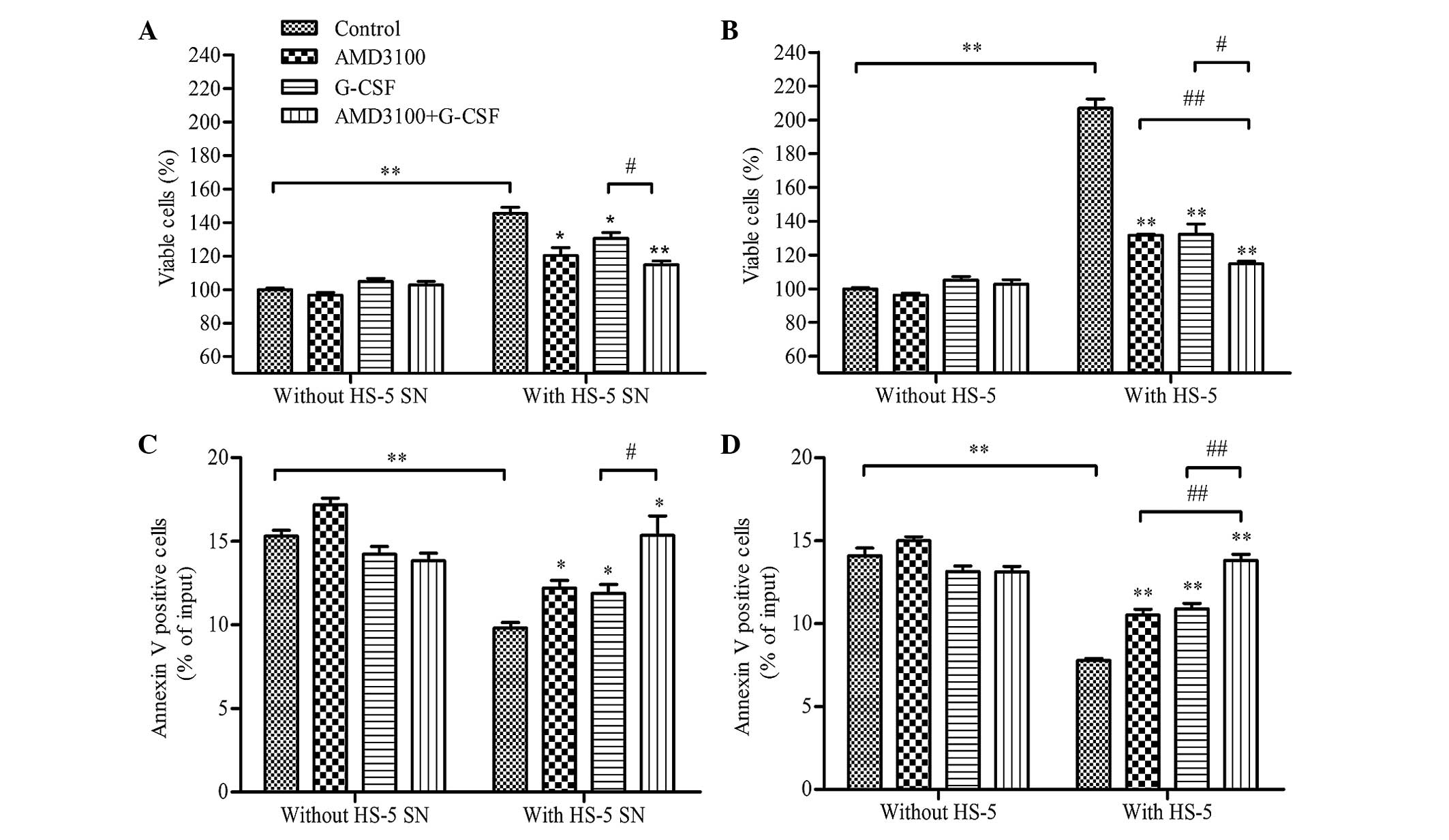
HS-5 cells protect HL-60 cells from
spontaneous apoptosis, whereas G-CSF reduces cell viability and
restores apoptosis
In order to investigate the effect of G-CSF on HL-60
cells in diverse microenvironments, HL-60 cells were co-cultured
with HS-5 supernatant (indirect contact), HS-5 cells (direct
contact) or medium alone (control), following pretreatment with
AMD3100, G-CSF, G-CSF plus AMD3100 or medium (control), and cell
viability and apoptosis were analyzed after 48 h. A protective
effect was observed in the HL-60 cells co-cultured with the HS-5
supernatant; cell viability was significantly increased (P<0.01;
Fig. 3A), and apoptosis was
significantly reduced (P<0.01; Fig.
3B), as compared with the HL-60 cells co-cultured with medium
only. Notably, G-CSF and/or AMD3100 did not significantly effect
the viability (Fig. 3A) or apoptosis
(Fig. 3B) of HL-60 cells that had
been cultured with medium alone (without HS-5 supernatant or HS-5
cells). However, pretreatment with G-CSF and/or AMD3100
significantly reduced the viability (P<0.05) and increased the
apoptosis (P<0.05) of HL-60 cells co-cultured with HS-5
supernatant, with G-CSF plus AMD3100 demonstrating greater effects
on HL-60 cell viability and apoptosis than either agent alone
(P<0.05; Figs. 3A and B). These
results suggested that G-CSF and AMD3100 antagonize the protective
effect of HS-5 cells on HL-60 cells that become more sensitive to
apoptosis, which may be rely on interfering with the CXCR4/SDF-1α
axis.
Similar results were observed for the HL-60 cells
co-cultured with HS-5 cells (Figs. 3C
and D). However, cell viability was significantly enhanced
(206.98±9.62% vs. 145.51±6.30%; P<0.01) and apoptosis was
significantly reduced (7.79±0.10% vs. 9.80±0.60%; P<0.01) for
the HL-60 cells co-cultured with HS-5 cells, as compared with those
co-cultured with HS-5 supernatant (Figs.
3A and B).
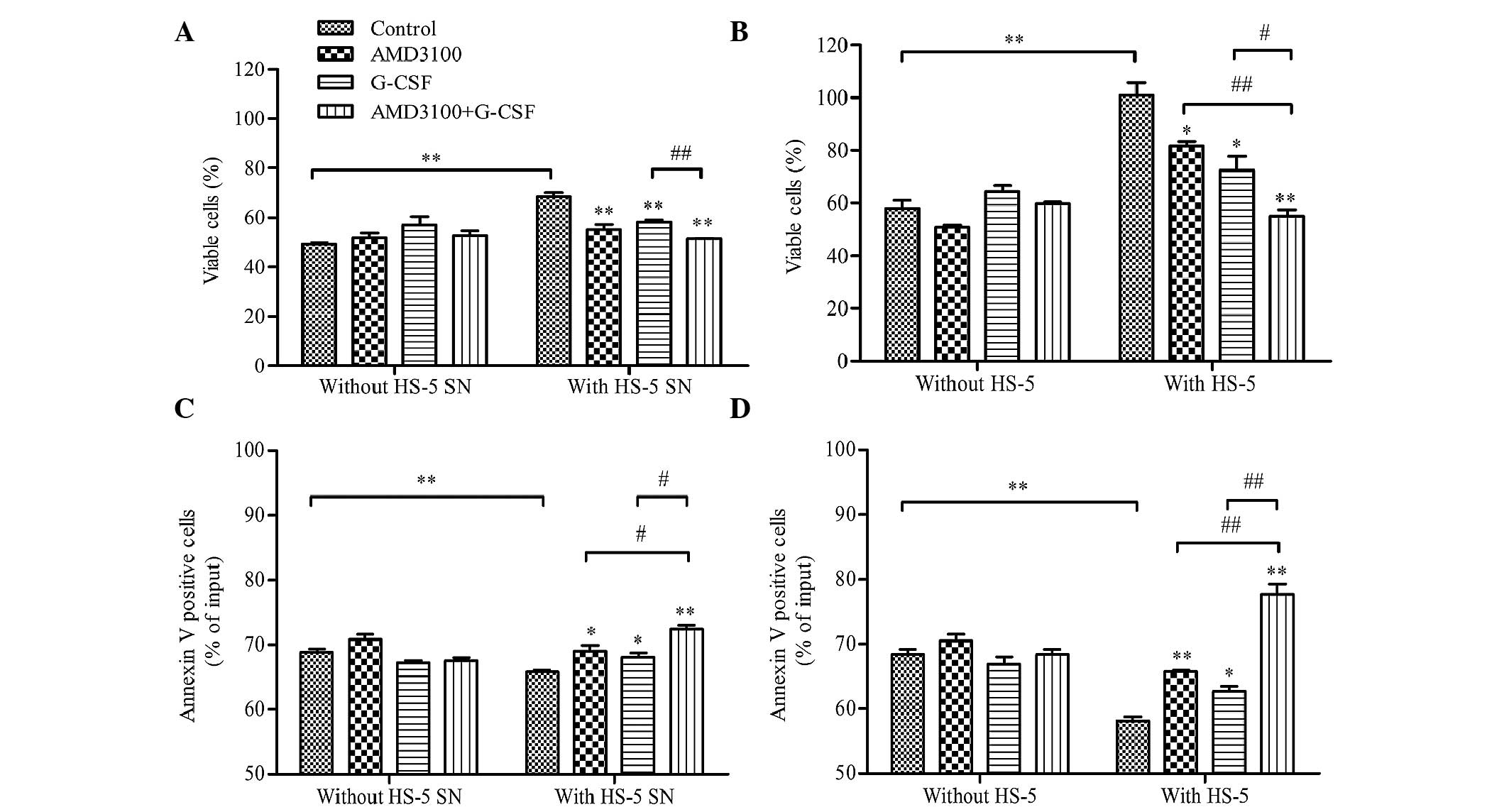
G-CSF sensitizes HL-60 cells to
drug-induced apoptosis in HL-60/HS-5 co-culture models
The effect of G-CSF on the drug-induced apoptosis of
HL-60 cells was evaluated using HL-60/HS-5 co-culture models. HL-60
cells were pre-treated with G-CSF, AMD3100, G-CSF plus AMD3100 or
medium (control), and then co-cultured with HS-5 supernatant, HS-5
cells or medium in the presence of cytarabine (0.05 µM). Following
48-h incubation, cell viability and apoptosis were analyzed. G-CSF
and AMD3100 did not influence cell viability or apoptosis when
HL-60 cells were co-cultured with medium alone in the presence of
cytarabine (Fig. 4). As compared
with the medium alone, co-culture with HS-5 supernatant or HS-5
cells protected HL-60 cells against cytarabine-induced apoptosis;
cell viability was significantly increased (P<0.01; Figs. 4A and C, respectively) and apoptosis
was significantly reduced (P<0.01, Figs. 4B and D, respectively). However,
pretreatment with G-CSF and/or AMD3100 significantly inhibited the
protective effects of the HS-5 supernatant or HS-5 cells, with
G-CSF plus AMD3100 exhibiting a greater effect on HL-60 cell
viability and apoptosis than either agent when used alone
(P<0.05; Fig. 4). In addition, a
more pronounced protective effect against cytarabine-induced
apoptosis was observed for the HL-60 cells co-cultured with HS-5
cells, as compared with the HS-5 supernatant (cell viability:
101.04±4.72% vs. 68.31±1.71%, P<0.01; apoptosis: 58.20±0.63% vs.
65.75±0.33%, P<0.05; Fig. 4).
These results suggest that the HS-5 supernatant and HS-5 cells
exert pro-proliferative and anti-apoptosis effects, which may be
increased when the leukemia cells are in physical contact with the
stromal cells. Furthermore, the results suggested that G-CSF and
AMD3100 may be able to restrict the stromal-based
microenvironment-mediated protective effects on leukemia cells.
G-CSF reduces the expression levels of
CXCR4 mRNA, surface protein and total protein, and increases the
expression of miR-146a in HL-60 cells
In functional assays, G-CSF and AMD3100 were able to
significantly inhibit the migration and adhesion of HL-60 cells,
and the protective effects of stromal cells. In order to elucidate
the association between the CXCR4/SDF-1α axis and the
stromal-mediated protective effects, the ability of G-CSF to alter
the expression levels of CXCR4 was evaluated. Firstly, surface
CXCR4 expression was evaluated using a PE-conjugated anti-CXCR4
monoclonal antibody and fluorescence-activated cell sorting. Mean
fluorescence intensity ratios of cells pre-treated with G-CSF
(9.87±0.45), AMD3100 (7.89±0.13) or G-CSF plus AMD3100 (2.88±0.17)
were significantly reduced, as compared with the control group
(14.29±0.32; P<0.01). Furthermore, combined treatment with G-CSF
and AMD3100 resulted in a greater reduction than when either agent
was used alone (P<0.01; Fig. 5A).
Next, the effect of G-CSF on the total CXCR4 content of HL-60 cells
was assessed by western blotting, since CXCR4 is predominantly an
intracellular protein (27). There
was a marked reduction of the 39-kDa CXCR4-associated band in the
HL-60 cells pretreated with G-CSF or G-CSF plus AMD3100, as
compared with the control group, whereas AMD3100 did not affect
CXCR4 protein expression (Fig. 5B).
Subsequently, the expression levels of CXCR4 mRNA and miR-146a were
analyzed by RT-qPCR. G-CSF significantly reduced CXCR4 mRNA
expression (P<0.01) and increased miR-146a expression
(P<0.01), as compared with the control; whereas AMD3100
treatment had no significant effect (Figs. 5C and D). In addition, a highly
inverse correlation was observed between the expression levels of
CXCR4 mRNA and miR-146a, which is indicative of
post-transcriptional inhibition of gene expression.
Overall, these results suggest that G-CSF inhibits
HL-60 cell migration and adhesion to HS-5 cells, and sensitizes
them to spontaneous or drug-induced apoptosis by disturbing the
CXCR4/SDF-1α axis via miR-146a upregulation.
Discussion
Substantial progress in the treatment of AML has
been achieved; in particular, the CAG regimen has demonstrated
great success in the treatment of elderly, relapsed/refractory and
secondary AML patients (1,28). Previous in vitro and in
vivo studies have highlighted the importance of G-CSF (29–31),
demonstrating that G-CSF has a key role in the CAG regimen via cell
cycle synchronization and cell mobilization. We suggest a novel
hypothesis that G-CSF may weaken the protection of stromal cells
and promote the apoptosis of leukemia cells. To the best of our
knowledge, no direct evidence in support of this has been
reported.
Spinello et al (32) demonstrated that patients with M4/M5
subtypes of AML had the highest levels of CXCR4 protein expression,
those with M3 had the second highest, and those with M1/M2 had the
lowest. Therefore, the present study selected a common human
leukemia cell line, HL-60, which is thought to be a cell line of
the AML-M2 subtype, and co-cultured it with the HS-5 human
BM/stromal cell line to imitate the interactions between stromal
cells and leukemia cells in vitro (33). AMD3100, which is a CXCR4 inhibitor,
has been used in humans for >10 years as a HSC mobilizing agent
(34). Previous studies have
demonstrated that AMD3100 exerts an inhibitory effect on the
CXCR4/SDF-1α axis (12,15,21,35);
thus AMD3100 was used as a positive control in the present
study.
Homing and retention in the BM are key protective
mechanisms for cells to escape drug-induced apoptosis and are
predominantly dependent on the CXCR4/SDF-1α axis (14). Therefore, the present study
investigated the effect of G-CSF on cell migration and adhesion,
which partially reflect homing and retention, respectively. The
results demonstrated that G-CSF significantly decreased the
migration and adhesion of HL-60 cells to HS-5 cells, which was
consistent with a previous study, in which a similar inhibitory
effect was reported for AMD3100 (35). In addition, the present study
demonstrated that G-CSF and AMD3100 had a greater inhibitory effect
on cell migration than on cell adhesion, which may be due to the
fact that cell adhesion involves numerous adhesion molecules,
whereas cell migration is predominantly dependent on the
CXCR4/SDF-1α axis (36). Although
cell adhesion in this assay did not only reflect CXCR4/SDF-1α
interactions, but also was dependent on contributions from other
molecules induced by CXCR4 activation, these results still provide
evidence that G-CSF may reduce functional CXCR4 levels in myeloid
cells.
Viability and apoptosis assays performed in the
present study demonstrated that co-culture with HS-5 supernatant
and HS-5 cells was able to protected HL-60 cells against
spontaneous or drug-induced apoptosis. Notably, a greater
protective effect was observed when HL-60 cells were co-cultured
with HS-5 cells (direct contact), as compared with when they were
co-cultured with HS-5 supernatant (indirect contact). These results
suggested that the protective effects of stromal cells were
predominantly dependent on physical contact, although soluble
factors were also involved. Furthermore, G-CSF decreased the
viability and promoted the apoptosis of HL-60 cells in the presence
or absence of cytarabine, although it was unable to affect the
viability and apoptosis of HL-60 cells cultured with medium alone.
Similar results were observed for AMD3100. These results suggested
that G-CSF and AMD3100 affected the survival and apoptosis of HL-60
cells by disrupting the interactions between HL-60 and HS-5 cells,
potentially via the CXCR4/SDF-1α axis, not as a result of their
toxicity. In addition, to the best of our knowledge, the present
study is the first to report synergistic effects for G-CSF and
AMD3100 on cell migration, adhesion, survival and apoptosis in
vitro, although previous studies have demonstrated their
synergistic effect in the mobilization of HSCs (37,38).
In functional assays, the present study demonstrated
that G-CSF was able to disrupt HS-5/HL-60 cell cross-talk by
interfering with the migration and adhesion of HL-60 cells to HS-5
cells, potentially via inhibition of the CXCR4/SDF-1α axis. In
order to further elucidate the involvement of the CXCR4/SDF-1α
axis, the ability of G-CSF to reduce the expression levels of
surface CXCR4 protein, total CXCR4 protein and CXCR4 mRNA in HL-60
cells was investigated. Notably, the surface expression of CXCR4
was markedly downregulated in the HL-60 cells pre-treated with
G-CSF, AMD3100 or with G-CSF plus AMD3100, and similar results were
observed for the total CXCR4 protein using western blotting.
Notably, the expression levels of surface CXCR4 protein were
markedly more decreased in the AMD3100 group, as compared with the
G-CSF group; whereas total CXCR4 protein expression was markedly
decreased in the HL-60 cells that had been pre-treated with G-CSF,
but not in those pre-treated with AMD3100. In 2011, Spinello et
al (32) reported that acute
treatment (1–4 h) with 10 µg AMD3100 elicited rapid downregulation
of surface CXCR4 expression, without any significant modulation of
CXCR4 mRNA and total protein expression levels. Therefore, the
authors of the present study hypothesized that these differences
may be due to differences in the mechanisms used by G-CSF and
AMD3100 to downregulate CXCR4 expression. AMD3100 may have
decreased CXCR4 surface expression via receptor internalization,
whereas G-CSF may have inhibited CXCR4 expression via translational
repression. Analysis of the expression levels of CXCR4 mRNA and
miR-146a supported this hypothesis. G-CSF significantly upregulated
miR-146a expression levels and downregulated CXCR4 mRNA expression
levels, whereas AMD3100 did not. To the best of our knowledge, the
present study is the first to demonstrate that G-CSF and AMD3100
may utilize different mechanisms in the downregulation of CXCR4
expression and exhibit synergistic anti-leukemia activity in
vitro.
In conclusion, the present study demonstrated that
G-CSF was able to overcome stromal-mediated drug resistance in the
HL-60 cell line by interfering with the CXCR4/SDF-1α axis, likely
via the upregulation of miR-146a expression in order to reduce
CXCR4 expression. These results suggested that there may be broader
prospects for the clinical application of G-CSF, as well as its use
as a mobilization agent.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81270626).
References
|
1
|
Zhu X, Ma Y and Liu D: Novel agents and
regiments for acute myeloid leukemia: 2009 ASH annual meeting
highlights. J Hematol Oncol. 3:172010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Estey EH: Acute myeloid leukemia: 2012
update on diagnosis, risk stratification and management. Am J
Hematol. 87:89–99. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rowe JM and Tallman MS: How I treat acute
myeloid leukemia. Blood. 116:3147–3156. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gregory TK, Wald D, Chen Y, Vermaat JM,
Xiong Y and Tse W: Molecular prognostic markers for adult acute
myeloid leukemia with normal cytogenetics. J Hematol Oncol.
2:232009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Juliusson G, Antunovic P, Derolf A,
Lehmann S, Mollgard L, Stockelberg D, Tidefelt U, Wahlin A and
Hoglund M: Age and acute myeloid leukemia: Real world data on
decision to treat and outcomes from the Swedish Acute Leukemia
Registry. Blood. 113:4179–4187. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yamada K, Furusawa S, Saito K, Waga K,
Koike T, Arimura H, Aoyagi A, Yamato H, Sakuma H, Tsunogake S, et
al: Concurrent use of granulocyte colony-stimulating factor with
low-dose cytosine arabinoside and aclarubicin for previously
treated acute myelogenous leukemia: A pilot study. Leukemia.
9:10–14. 1995.PubMed/NCBI
|
|
7
|
Wei G, Ni W, Chiao JW, Cai Z, Huang H and
Liu D: A meta-analysis of CAG (cytarabine, aclarubicin, G-CSF)
regimen for the treatment of 1029 patients with acute myeloid
leukemia and myelodysplastic syndrome. J Hematol Oncol. 4:462011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Collins RJ, Verschuer LA, Harmon BV,
Prentice RL, Pope JH and Kerr JF: Spontaneous programmed death
(apoptosis) of B-chronic lymphocytic leukaemia cells following
their culture in vitro. Br J Haematol. 71:343–350. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nwajei F and Konopleva M: The bone marrow
microenvironment as niche retreats for hematopoietic and leukemic
stem cells. Adv Hematol. 2013:9539822013.PubMed/NCBI
|
|
10
|
Mèndez-Ferrer S, Michurina TV, Ferraro F,
Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma'ayan A,
Enikolopov GN and Frenette PS: Mesenchymal and haematopoietic stem
cells form a unique bone marrow niche. Nature. 466:829–834. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Caligaris-Cappio F: Role of the
microenvironment in chronic lymphocytic leukaemia. Br J Haematol.
123:380–388. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Azab AK, Runnels JM, Pitsillides C, Moreau
AS, Azab F, Leleu X, Jia X, Wright R, Ospina B, Carlson AL, et al:
CXCR4 inhibitor AMD3100 disrupts the interaction of multiple
myeloma cells with the bone marrow microenvironment and enhances
their sensitivity to therapy. Blood. 113:4341–4351. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yin T and Li L: The stem cell niches in
bone. J Clin Invest. 116:1195–1201. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zeng Z, Shi YX, Samudio IJ, Wang RY, Ling
X, Frolova O, Levis M, Rubin JB, Negrin RR, Estey EH, et al:
Targeting the leukemia microenvironment by CXCR4 inhibition
overcomes resistance to kinase inhibitors and chemotherapy in AML.
Blood. 113:6215–6224. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tavor S, Eisenbach M, Jacob-Hirsch J,
Golan T, Petit I, Benzion K, Kay S, Baron S, Amariglio N, Deutsch
V, et al: The CXCR4 antagonist AMD3100 impairs survival of human
AML cells and induces their differentiation. Leukemia.
22:2151–5158. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sison EA, Rau RE, McIntyre E, Li L, Small
D and Brown P: MLL-rearranged acute lymphoblastic leukaemia stem
cell interactions with bone marrow stroma promote survival and
therapeutic resistance that can be overcome with CXCR4 antagonism.
Br J Haematol. 160:785–797. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Busillo JM and Benovic JL: Regulation of
CXCR4 signaling. Biochim Biophys Acta. 1768:952–963. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vianello F, Villanova F, Tisato V, Lymperi
S, Ho KK, Gomes AR, Marin D, Bonnet D, Apperley J, Lam EW and Dazzi
F: Bone marrow mesenchymal stromal cells non-selectively protect
chronic myeloid leukemia cells from imatinib-induced apoptosis via
the CXCR4/CXCL12 axis. Haematologica. 95:1081–1089. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Burger JA and Kipps TJ: CXCR4: A key
receptor in the crosstalk between tumor cells and their
microenvironment. Blood. 107:1761–1767. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
He L and Hannon GJ: MicroRNAs: Small RNAs
with a big role in gene regulation. Nat Rev Genet. 5:522–531. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen Y, Stamatoyannopoulos G and Song CZ:
Down-regulation of CXCR4 by inducible small interfering RNA
inhibits breast cancer cell invasion in vitro. Cancer Res.
63:4801–4804. 2003.PubMed/NCBI
|
|
22
|
Labbaye C, Spinello I, Quaranta MT, Pelosi
E, Pasquini L, Petrucci E, Biffoni M, Nuzzolo ER, Billi M, Foà R,
et al: A three-step pathway comprising PLZF/miR-146a/CXCR4 controls
megakaryopoiesis. Nat Cell Biol. 10:788–801. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim HK, De La Luz Sierra M, Williams CK,
Gulino AV and Tosato G: G-CSF down-regulation of CXCR4 expression
identified as a mechanism for mobilization of myeloid cells. Blood.
108:812–820. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
De La Luz Sierra M, Gasperini P,
McCorimick PJ, Zhu J and Tosato G: Transcription factor Gfi-1
induced by G-CSF is a negative regulator of CXCR4 in myeloid cells.
Blood. 110:2276–2285. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Donahue RE, Jin P, Bonifacino AC, Metzger
ME, Ren J, Wang E and Stroncek DF: Plerixafor (AMD3100) and
granulocyte colony-stimulating factor (G-CSF) mobilize different
CD34+ cell populations based on global gene and microRNA expression
signatures. Blood. 114:2530–2541. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Förster R, Kremmer E, Schubel A, Breitfeld
D, Kleinschmidt A, Nerl C, Bernhardt G and Lipp M: Intracellular
and surface expression of the HIV-1 coreceptor CXCR4/fusin on
various leukocyte subsets: Rapid internalization and recycling upon
activation. J Immunol. 160:1522–1531. 1998.PubMed/NCBI
|
|
28
|
Fernandez HF, Sun Z, Yao X, Litzow MR,
Luger SM, Paietta EM, Racevskis J, Dewald GW, Ketterling RP,
Bennett JM, et al: Anthracycline dose intensification in acute
myeloid leukemia. N Engl J Med. 361:1249–1259. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bai A, Kojima H, Hori M, Nara N, Komeno T,
Hasegawa Y, Ninomiya H, Abe T and Nagasawa T: Priming with G-CSF
effectively enhances low-dose Ara-C-induced in vivo apoptosis in
myeloid leukemia cells. Exp Hematol. 27:259–265. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tafuri A and Andreeff M: Kinetic rationale
for cytokine-induced recruitment of myeloblastic leukemia followed
by cycle-specific chemotherapy in vitro. Leukemia. 4:826–834.
1990.PubMed/NCBI
|
|
31
|
Ferrero D, Carlesso N, Pregno P, Gallo E
and Pileri A: Self-renewal inhibition of acute myeloid leukemia
clonogenic cells by biological inducers of differentiation.
Leukemia. 6:100–106. 1992.PubMed/NCBI
|
|
32
|
Spinello I, Quaranta MT, Riccioni R, Riti
V, Pasquini L, Boe A, Pelosi E, Vitale A, Foà R, Testa U and
Labbaye C: MicroRNA-146a and AMD3100, two ways to control CXCR4
expression in acute myeloid leukemias. Blood Cancer J. 1:e262011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dalton WT Jr, Ahearn MJ, McCredie KB,
Freireich EJ, Stass SA and Trujillo JM: HL-60 cell line was derived
from a patient with FAB-M2 and not FAB-M3. Blood. 71:242–247.
1988.PubMed/NCBI
|
|
34
|
Hendrix CW, Flexner C, MacFarland RT,
Giandomenico C, Fuchs EJ, Redpath E, Bridger G and Henson GW:
Pharmacokinetics and safety of AMD-3100, a novel antagonist of the
CXCR-4 chemokine receptor, in human volunteers. Antimicrob Agents
Chemother. 44:1667–1673. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Stamatopoulos B, Meuleman N, De Bruyn C,
Pieters K, Mineur P, Le Roy C, Saint-Georges S, Varin-Blank N,
Cymbalista F, Bron D and Lagneaux L: AMD3100 disrupts the
cross-talk between chronic lymphocytic leukemia cells and a
mesenchymal stromal or nurse-like cell-based microenvironment:
Pre-clinical evidence for its association with chronic lymphocytic
leukemia treatments. Haematologica. 97:608–615. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Campbell JJ, Qin S, Bacon KB, Mackay CR
and Butcher EC: Biology of chemokine and classical chemoattractant
receptors: differential requirements for adhesion-triggering versus
chemotactic responses in lymphoid cells. J Cell Biol. 134:255–266.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Flomenberg N, Devine SM, DiPersio JF,
Liesveld JL, McCarty JM, Rowley SD, Vesole DH, Badel K and Calandra
G: The use of AMD3100 plus G-CSF for autologuos hematopoietic
progentior cell mobilization is superior to G-CSF alone. Blood.
106:1867–1874. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Winkler IG, Pettit AR, Raggatt LJ,
Jacobsen RN, Forristal CE, Barbier V, Nowlan B, Cisterne A, Bendall
LJ, Sims NA and Lévesque JP: Hematopoietic stem cell mobilizing
agents G-CSF, cyclophosphamide or AMD3100 have distinct mechanisms
of action on bone marrow HSC niches and bone formation. Leukemia.
26:1594–1601. 2012. View Article : Google Scholar : PubMed/NCBI
|