Introduction
Acute kidney injury (AKI) is a common disorder
(complicating 3–50% of hospital admissions) that is expensive to
manage, prolongs the hospitalization of patients and is associated
with increased mortality (1). AKI is
characterized by an acute reduction in kidney function, exhibited
as an increase in serum creatinine levels and reduction in urine
output (2). AKI has been found to be
associated with a high morbidity and mortality in up to 22% of
hospitalized patients (3). AKI
commonly occurs in patients with acute illness and is a
long-recognized complication of cardiac surgery (4). In its most severe form, AKI increases
the odds of operative mortality by 3–8 fold (5,6). A
number of different events are known to cause AKI, including
ischemia, vasoconstrictive drugs, exposure to toxins, hypotension
linked to sepsis, and obstruction of the urinary tract, and this
condition may lead to various complications, including metabolic
acidosis, high potassium levels, uremia, changes in body fluid
balance, and damage to and failure of various organs (7). Since the mechanisms underlying AKI are
unclear, no specific therapy is currently available to prevent or
treat it (8).
Kidney injury molecule-1 (Kim-1) is a transmembrane
glycoprotein that typically cannot be detected in the healthy
kidney, but which is upregulated and excreted in the urine during
the early stages of ischemic or nephrotoxicity-induced proximal
tubule injury (9,10). Kim-1 is a marker of the majority of
proteinuric, toxic and ischemia-associated kidney diseases. Kim-1
has gained increasing interest due to its potential role in the
modulation of tubular damage and repair (11). Previous studies have suggested that
Kim-1 may be involved in the differentiation and proliferation of
proximal tubule endothelial cells, and may participate in the
restoration of renal tubular epithelium tissue, as well as
maintaining its functional integrity (11–13).
However, at present, the upstream molecular mechanism underlying
the dysregulation of Kim-1 expression in AKI is unclear.
Aldosterone (Aldo) is an important mediator of the
renin-angiotensin (Ang)-Aldo system (RAAS) and has a pivotal role
in the regulation of salt and extracellular fluid metabolism. In
addition, Aldo exerts non-hemodynamic effects associated with end
organ damage and renal injury (14).
These detrimental effects of Aldo are reported to be independent of
the RAAS (14). A previous study
demonstrated that RAAS induced Kim-1 expression in proximal tubule
endothelial cells, leading to kidney injury, whereas treatment with
an Aldo receptor antagonist was able to decrease Kim-1 expression
and attenuate kidney injury (15).
These results suggest that Aldo is associated with the expression
of Kim-1; however, studies investigating the molecular mechanisms
underlying the role of Aldo in Kim-1 expression are lacking.
MicroRNAs (miRNAs) are endogenous RNA molecules,
usually 18–25 nucleotides in length, that regulate gene expression
at the post-transcriptional level. miRNAs typically target the
3′-untranslated regions (UTRs) of mRNA, and since miRNA target
motifs do not require complete homology, there may be multiple mRNA
targets for each miRNA (16).
miRNAs are required for normal development, cell
growth, differentiation, apoptosis and the regulation of numerous
other biological processes in mammals (17). Since they participate in almost every
cellular process, their dysregulation has been associated with the
development of numerous pathologies, including kidney diseases
(18).
miRNAs have been indicated to be key regulators of
normal kidney function and development, and are the basis of
several renal diseases (18). AKI
has been associated with aberrantly expressed miRNAs in several
studies; circulating miR-210 predicted improved rates of survival
in patients with AKI (19), whereas
plasma levels of miR-21 were associated with the progression of AKI
(20), and the levels of miR-494
were markedly increased in the urine of patients with AKI (21). Furthermore, some miRNAs have been
reported to be novel biomarkers and therapeutic candidates for AKI,
including miR-714, miR-1188, miR-1897-3p and miR-182-5p (22,23).
However, whether miRNA-mediated dysregulation of Kim-1 occurs has
yet to be elucidated.
The present study aimed to investigate the
association between the Aldo-induced upregulation of Kim-1
expression and the onset of necrotic and apoptotic cell death in
proximal tubule endothelial cells, as well as the potential
molecular mechanism underlying the role of miRNAs in AKI.
Materials and methods
General animal procedures
A total of 24 male CD-1 mice (weight, 25–35 g; aged
6–8 weeks; Xiangya Hospital of Central South University, Changsha,
China) were used for the experiments. The mice were maintained
under routine vivarium conditions (temperature, 18–26°C; humidity,
40–70%; light/dark cycle, 10 h/14 h) with ad libitum access
to food and water throughout. All procedures were performed in
accordance with the animal care guidelines at the Xiangya Hospital
of Central South University. Ethical approval was provided by the
Ethics Committee of the Center for Scientific Research with Animal
Models of Central South University. All surgeries were conducted
under deep anesthesia, achieved by the intraperitoneal injection of
pentobarbital sodium (2–3 mg/mouse; Beijing Solarbio Science &
Technology, Co., Ltd., Beijing, China). During anesthesia and
recovery, the body temperature of the mice was maintained at
36–37°C using heating lamps during surgery, or by maintaining the
mice in a temperature-controlled apparatus during recovery. The
mice were divided into four groups, with 6 mice in each group, as
follows: Control (con), sham, ischemia/reperfusion (I/R) and I/R +
spironolactone (SPIR) groups. The mice in the I/R + SPIR group were
administered spironolactone (20 mg/kg/day; Sigma-Aldrich, St.
Louis, MO, USA) by gavage 5 days prior to model construction.
Renal I/R model construction
Mice models of renal I/R were established, as
described previously (24). Briefly,
mice were subjected to a midline abdominal incision, the left renal
pedicle was identified and a smooth vascular clamp was applied to
it. Following 15 min of ischemia, reperfusion was confirmed by the
attenuation of organ cyanosis. The right kidney was left
unperturbed. Following recovery from anesthesia, the mice were
returned to their cages. After 48 h of reperfusion, the mice were
reanesthetized, the abdominal incisions were opened and
contralateral kidneys were removed. The mice in the sham group were
subjected to a midline abdominal incision; however, the kidneys
were left unperturbed.
Immunohistochemistry
Renal tissues were obtained, fixed with 4%
formaldehyde solution overnight, dehydrated using an alcohol
gradient, cleared using xylene and paraffin-embedded. Subsequently,
the paraffin-embedded tissue blocks were sliced into 8 nm sections
using a microtome, which were then mounted onto anti-shedding glass
slides. The slides were baked in an oven at 55°C for 2 h, after
which the sections were deparaffinized and rehydrated, prior to
antigen retrieval using sodium citrate. The sections were incubated
with rabbit anti-Kim-1 polyclonal antibody (cat. no. ABF199; EMD
Millipore, Billerica, MA, USA) or normal rabbit immunoglobulin G
(GenWay Biotech, Inc., San Diego, CA, USA) as a negative control
overnight at 4°C. After this, the sections were treated with
Universal Immuno-peroxidase Polymer Anti-Mouse/Rabbit
Immunohistochemical Staining reagent (PV-8000; ZSGB-Bio, Beijing,
China) according to the manufacturer's protocol. Immunostaining was
performed using diaminobenzidine (Zhongshan Jinqiao Biological
Technology Co., Ltd., Beijing, China) and the stained tissues were
examined using the Moticam 3000 system (Motic, Causeway Bay, Hong
Kong). Image Pro-Plus software, version 6.0 (Media Cybernetics,
Inc., Rockville, MD, USA) was used to quantify the mean density of
Kim-1 staining, according to the manufacturer's protocol.
Detection of Aldo by radioimmunoassay
(RIA)
Urine samples were collected from the mice after 48
h of reperfusion for the analysis of Aldo by RIA. Prior to
analysis, urine samples (50 µl) were centrifuged at 3,000 × g for
10 min, after which the supernatant was treated with HCl. Urinary
Aldo levels in the supernatant were determined using an Iodine-125
Aldosterone Radioimmunoasssay kit (Shanghai Rongbai Biological
Technology, Co., Ltd., Shanghai, China), according to the
manufacturer's protocol. The assay tubes were centrifuged at 1,000
× g for 45 min, after which the supernatant was mixed with 3 ml
liquid scintillation fluid (Beckman Coulter, Inc., Brea, CA, USA)
prior to counting the radioactivity in an automated β-counter.
Duplicate standard curves with five points ranging from 3 to 800 pg
Aldo were obtained. Aldo levels in the urine samples were
determined by comparison with the standard curve.
Cell culture and treatment
NRK-52E rat proximal tubular cells (American Type
Culture Collection, Manassas, VA, USA) were cultured in Dulbecco's
modified Eagle's medium (Sigma-Aldrich) supplemented with 10% fetal
bovine serum, 2% penicillin-streptomycin (both Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), 1% HEPES buffer, 2 g sodium
bicarbonate and 2 mM L-glutamine, in 5% CO2 at 37°C,
according to a previous study (25).
Subsequently, the cells were incubated for 12 h in an environment
containing 94% N2, 5% CO2 and 1% O2 to
simulate hypoxia, followed by reoxygenation for 6 h to generate
hypoxia/reoxygenation (H/R) cell models. Other cells were treated
with 0.1 µmol/l antimycin A (Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA) for 24 h at 30°C, to create a different fibrotic
cell model.
In the subsequent drug treatment experiments, the
cells were serum-starved for 24 h, and then treated with either 50
nM Aldo or 100 nM spironolactone (both Sigma-Aldrich) for 1 h at
30°C. In addition, when the cells were treated with both
spironolactone and Aldo, the cells were pretreated with 100 nM
spironolactone for 1 h prior to the addition of 50 nM Aldo for 1
h.
Cell transfection and dual luciferase
reporter assay
In order to analyze the function of miR-203 in AKI,
pre-miR-203 or pre-control (FulenGen Co., Ltd., Guangzhou, China)
was transfected into NRK-52E cells using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. The 3′-UTR of Kim-1 (NM_173149.2)
containing the Rattus norvegicus (rno)-miR-203 binding sites
or its corresponding mutated sequence was cloned into the
psi-CHECK™-2 luciferase reporter vector (Promega Corporation,
Madison, WI, USA) downstream of the Renilla luciferase, generating
Kim-1–3′-UTR and Kim-1-Mut 3′-UTR vectors, respectively.
Subsequently, NRK-52E cells were co-transfected with the
recombinant reporter constructs and rno-miR-203 mimics, the
rno-miR-203 inhibitor, negative control (NC) or NC inhibitor
(GeneCopoeia, Guangzhou, China) using Lipofectamine 2000.
Luciferase activity was determined after 48 h using the
Dual-Glo® Luciferase Assay system (Promega Corporation)
and an LD 400 Luminometer (Beckman Coulter, Inc.). Data are
presented as the ratio of experimental (Renilla) luciferase to
control (firefly) luciferase.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol. The relative
expression levels of rno-miR-203 were determined by RT-qPCR using
the mirVana™ qRT-PCR microRNA Detection kit (Ambion; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
Specific primer sets for rno-miR-203 (cat. no. RmiRQP0305) and U6
(used as an internal reference; cat. no. HmiRQP9003) were obtained
from GeneCopoeia (Rockville, MD, USA). The relative mRNA expression
levels of Kim-1 were detected using the standard SYBR Green RT-PCR
kit (Takara Bio, Inc., Otsu, Japan), according to the
manufacturer's protocol. The specific primer pairs were as follows:
Kim-1 sense, 5′-AAGGGCTTCTATGTTGGC-3′ and antisense,
5′-CCTCTGGGACTCATTCTG-3′; and β-actin as an internal control,
sense, 5′-CCCATCTATGAGGGTTACGC-3′ and antisense,
5′-TTTAATGTCACGCACGATTTC-3′ (Takara Bio, Inc.). The PCR analysis
was conducted using an ABI 7300 Real-Time PCR system (Thermo Fisher
Scientific, Inc.) with the following cycling conditions: A single
cycle at 95°C for 3 min, followed by 35 cycles of 95°C for 10 sec
and 58°C for 30 sec. The relative expression levels of Kim-1 mRNA
or rno-miR-203 were quantified using GraphPad Prism 4.0 software
(GraphPad Software, San Diego, CA, USA) and the 2−ΔΔCq
method (26).
Western blotting
Tissues or cells were solubilized in cold
radioimmunoprecipitation assay lysis buffer (Auragene Bioscience,
Changsha, China) and protein concentrations were determined using a
bicinchoninic acid protein assay kit. The proteins (10 µg) were
separated by 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and then transferred to polyvinylidene difluoride
membranes. The membranes were blocked with 10% non-fat dried milk
in phosphate-buffered saline (PBS) containing 0.01% Tween-20 for 3
h at room temperature and then incubated overnight at 4°C with
mouse anti-Kim-1 monoclonal antibody (1:1,000; ab66062; Abcam,
Cambridge, MA, USA), mouse anti-Ang II monoclonal antibody
(1:1,000; ab9391; Abcam), rabbit anti-caspase-8 polyclonal antibody
(1:1,000; AM2288; Abzoom Biolabs, Inc., Dallas, TX, USA), rabbit
anti-caspase-9 polyclonal antibody (1:1,000; AM2289; Abzoom
Biolabs, Inc.), rabbit anti-cleaved poly (ADP-ribose) polymerase
(PARP) polyclonal antibody (1:1,500; sc-23461; Santa Cruz
Biotechnology, Inc.) and mouse anti-β-actin monoclonal antibody
(1:2,000; LCA01; Auragene Bioscience). Following subsequent
incubation with horseradish peroxidase-conjugated goat anti-rabbit
(1:3,000: 111-035-008; Jackson ImmunoResearch Laboratories, Inc.,
West Grove, PA, USA) and anti-mouse (1:15,000; SA001; Auragene
Bioscience) secondary antibodies for 1 h at room temperature, the
immune complexes were detected using an ECL kit (P001WB-1; Auragene
Bioscience). Data was analyzed by densitometry using Image-Pro plus
software, version 6.0. Each condition was tested in triplicate, and
expression levels were normalized to β-actin expression.
Methylation-specific PCR (MSP)
Genomic DNA was extracted from cells or tissues
using a Genomic DNA Extraction kit (Takara Biotechnology Co., Ltd.,
Dalian, China). Genomic DNA (1 µg) was modified with bisulfite
using the Epitect Bisulfite kit (Qiagen, Inc., Valencia, CA, USA)
according to the manufacturer's protocol, and eluted with 40 ml
elution buffer. The modified DNA (2 ml) was then used as the
template in a bisulfite PCR reaction. MSP was conducted on the
bisulfate-treated DNA using the EZ DNA Methylation-Gold™ kit (Zymo
Research Corporation, Irvine, CA, USA), according to the
manufacturer's protocol. Methylation-specific primers for the
rno-miR-203 promoter were designed using the MethPrimer program
(http://www.urogene.org/methprimer/).
Bisulfite-converted genomic DNA was amplified by PCR using
methylation-specific primers and the SYBR Green reaction mix
(Takara Bio, Inc.). The primers used were as follows: MSP primer
forward, 5′-GTCGGTGATTTAGGGTTATTTTC-3′ and reverse,
5′-GACTACACTCCGTACGACGA-3′; and unmethylated-specific primer
forward, 5′-GGTTGGTGATTTAGGGTTATTTTT-3′, and reverse,
5′-TAACCCAACTACACTCCATACAACA-3′. Cycling was conducted using an ABI
7300 system, with cycling conditions of 94°C for 4 min; then 35
cycles of 94°C for 30 sec, 55°C for 30 sec and 72°C for 30 sec;
followed by 72°C for 5 min. The sample was then maintained at
4°C.
Flow cytometric analysis by annexin
V-fluorescein isothiocyanate (FITC) and propidium iodide (PI)
double-staining
The apoptosis of NRK-52E cells was analyzed by flow
cytometric analysis using the FITC Annexin V Apoptosis Detection
Kit I (BD Pharmingen, San Diego, CA, USA) and PI staining,
according to the manufacturer's protocols. Briefly, the cells were
washed twice with cold PBS and then resuspended in binding buffer.
Subsequently, 2×105 cells/ml were transferred to a 5-ml
tube containing 5 µl annexin V-FITC and 5 µl PI, followed by
incubation for 15 min at room temperature in the dark. Following
the addition of 400 µl binding buffer, the samples were analyzed by
flow cytometry (FACSCalibur; BD Biosciences, Franklin Lakes, NJ,
USA) within 1 h. Annexin V-positive and PI-negative cells were
defined as apoptotic. Data analysis was performed with ModFit LT
software, version 2.0 (Verity Software House, Inc., Topsham, ME,
USA).
Bioinformatics
Bioinformatics analysis was conducted using
Targetscan (http://www.targetscan.org/) and MicroRNA.org (http://www.microrna.org/microrna/getGeneForm.do)
software in order to predict the possibility that a direct target
relationship exists between Kim-1 and miR-203. The Li Lab
(http://www.urogene.org/) was used to perform the
CpG island prediction.
Statistical analysis
Data are presented as the mean ± standard deviation
of at least three independent experiments. Statistical analyses
were performed using SPSS 16.0 software (SPSS, Inc., Chicago, IL,
USA). Differences between groups were analyzed by one-way analysis
of variance, followed by Fisher's Least Significant Difference
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Expression levels of Kim-1 and levels
of Aldo in I/R mice models
In order to investigate Kim-1 expression in AKI
renal tissues, I/R mice models were established with or without
spironolactone feeding, and urine and renal tissues were collected
after 48 h. Immunohistochemical analysis was performed in order to
detect the expression of Kim-1 in the renal tissues of the various
groups and RT-qPCR was conducted to determine the relative mRNA
expression levels of Kim-1. Immunohistochemical analyses
demonstrated that the expression of Kim-1 was negligible in normal
renal tissues; however, it was significantly upregulated in the
renal tissues of the group subjected to I/R (Fig. 1A). The mRNA expression level of Kim-1
was markedly lower in the I/R mice treated with spironolactone
compared with that in the I/R group (Fig. 1B).
The Aldo levels in the urine of the mice were
detected using a RIA. The Aldo level in the urine of the mice in
the I/R group was significantly higher compared with that of the
control and sham groups (P<0.05; Fig.
1C); however, upon treatment of the I/R mice with
spironolactone (20 mg/kg/day), the Aldo level in the urine of the
mice was decreased, although it remained significantly higher than
the levels in the control and sham groups (P<0.05; Fig. 1C). These results suggest that
elevated Aldo levels may induce Kim-1 expression in the renal
tissues of I/R mice. In order to verify these results, western
blotting was performed to detect the protein expression levels of
Kim-1 and Ang II in the renal tissues from the various groups. The
protein expression levels of Kim-1 and Ang II were markedly
upregulated in the I/R group, as compared with the control and sham
groups (Fig. 1D).
Expression of Ang II and Kim-1 in
NRK-52E cell models
In order to establish H/R cell models, NRK-52E cells
were subjected to 94% N2, 5% CO2 and 1% O2 to
simulate hypoxia, and then reoxygenated. In addition, NRK-52E cells
were treated with 0.1 µmol/l antimycin A to obtain a fibrotic cell
model. RT-qPCR and western blotting were performed to detect the
mRNA and protein expression levels of Kim-1 and Ang II. The mRNA
and protein expression levels of Ang II and Kim-1 were
significantly upregulated in the H/R and antimycin A-treated groups
as compared with the control group (P<0.01; Fig. 2). However, in the cells that had been
pre-treated with the Aldo inhibitor spironolactone, the mRNA and
protein expression levels of Ang II and Kim-1 were significantly
reduced, as compared with those in the H/R and antimycin A-treated
groups (P<0.05), although they remained significantly elevated,
as compared with the levels in control cells (P<0.05; Fig. 2). These results suggest that Aldo may
have an important role in the upregulation of Kim-1 in H/R and
antimycin A-treated cell models.
Relative expression levels of
rno-miR-203 and its association with Kim-1 in NRK cells
Bioinformatics software has predicted that Kim-1 may
be a target gene of rno-miR-203. The present study aimed to
determine the molecular mechanisms underlying the Aldo-mediated
upregulation of Kim-1 expression in NRK-52E cells. qPCR was
performed to detect the levels of rno-miR-203 in the renal tissues
of the various I/R mouse models and in the cell groups. The levels
of rno-miR-203 were downregulated in the renal tissues of the I/R
mice, as compared with the control group (P<0.01; Fig. 3A). Conversely, the levels of
rno-miR-203 were significantly increased in the renal tissues of
the I/R mice treated with spironolactone, as compared with those in
the I/R group (P<0.05; Fig.
3A).
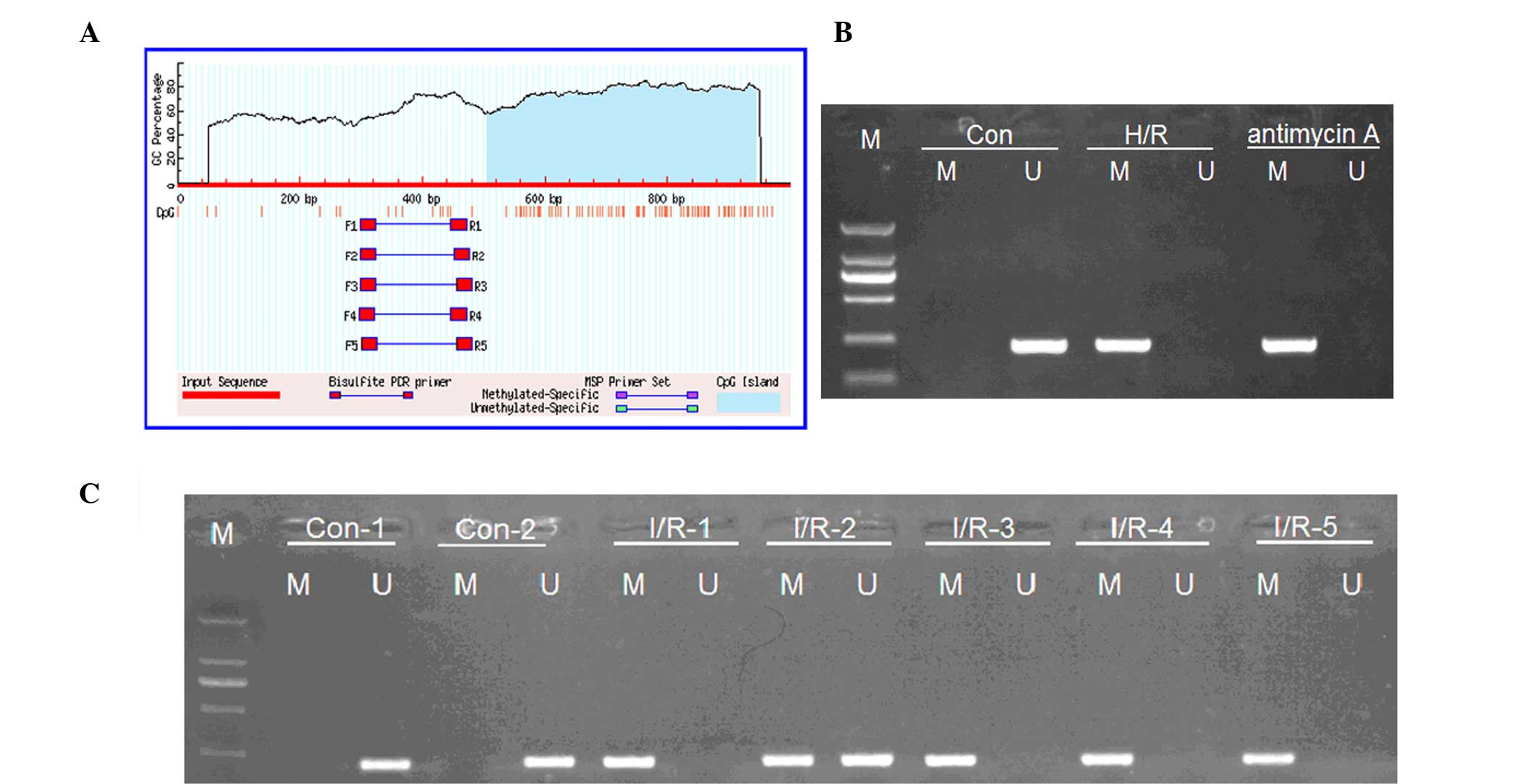
 | Figure 3.Kim-1 is a direct target of
rno-miR-203 in NRK-52E cells. Relative levels of rno-miR-203 in (A)
renal tissues of mice in the I/R and I/R + SPIR groups and (B) H/R-
and antimycin A-treated NRK cells with or without SPIR determined
by reverse transcription-quantitative polymerase chain reaction.
*P<0.05, **P<0.01 vs. the con group. (C) Dual luciferase
reporter assays were conducted to test the interaction of miR-203
and the 3′-UTR of Kim-1, using constructs containing the target
sequence (Kim-1–3′UTR) or mutated target sequence (Mut-Kim-1–3′UTR
Mut) cloned into psi-CHECK-2 luciferase reporter vectors and
transfected into cells. Data are presented as the mean ± standard
deviation (n=3). *P<0.05 vs. the Con group.
#P<0.05 between groups. Kim-1, kidney injury
molecule-1; miR, microRNA; I/R, ischemia/reperfusion; SPIR,
spironolactone; H/R, hypoxia/reoxygenation; con, control; Mut,
mutant; 3′-UTR, 3′-untranslated region; NC, negative control. |
The levels of rno-miR-203 were significantly
downregulated in the NRK-52E cells treated with H/R or antimycin A
as compared with those in normal cells (P<0.01; Fig. 3B), whereas the levels of rno-miR-203
were markedly increased in the H/R or antimycin A-treated cells
pre-treated with spironolactone, as compared with those in the
non-pre-treated cells (Fig. 3B).
These results suggest that Aldo may have a role in the
downregulation of rno-miR-203 in the renal tissues of animals with
AKI and in cell models.
In order to assess whether Kim-1 is a direct target
of rno-miR-203, luciferase reporter assays were conducted. The
Kim-1 3′-UTR fragment containing the rno-miR-203 binding site or a
mutated targeting sequence was cloned into a psi-CHECK-2 Dual
Luciferase Reporter vector. rno-miR-203 significantly inhibited the
luciferase activity in NRK-52E cells transfected with the
Kim-1–3′-UTR (P<0.05; Fig. 3C).
However, rno-miR-203 mimics did not suppress the luciferase
activity levels in NRK-52E cells transfected with Mut-Kim-1–3′-UTR
(Fig. 3C). These results suggest
that Kim-1 is a direct target of rno-miR-203 in NRK-52E cells.
Rno-miR-203 is hypermethylated in AKI
tissues and cells treated with H/R or antimycin A
The present study aimed to determine the mechanisms
underlying the Aldo-mediated downregulation of rno-miR-203 in AKI
tissues and cells. Bioinformatics software predicted that there is
a CpG island in the promoter of rno-miR-203 (Fig. 4A). Therefore, MSP was performed in
order to detect the methylation status of the rno-miR-203. MSP
demonstrated that both methylated and non-methylated rno-miR-203
were present in the renal tissues of the I/R mice (Fig. 4B), and the NRK-52E cells treated with
H/R or antimycin A. Conversely, only non-methylated rno-miR-203 was
detected in the control (Fig. 4C).
These results suggest that, when AKI occurs, the downregulation of
rno-miR-203 may be in part due to its methylation, which in turn
may promote the expression of its direct target, Kim-1.
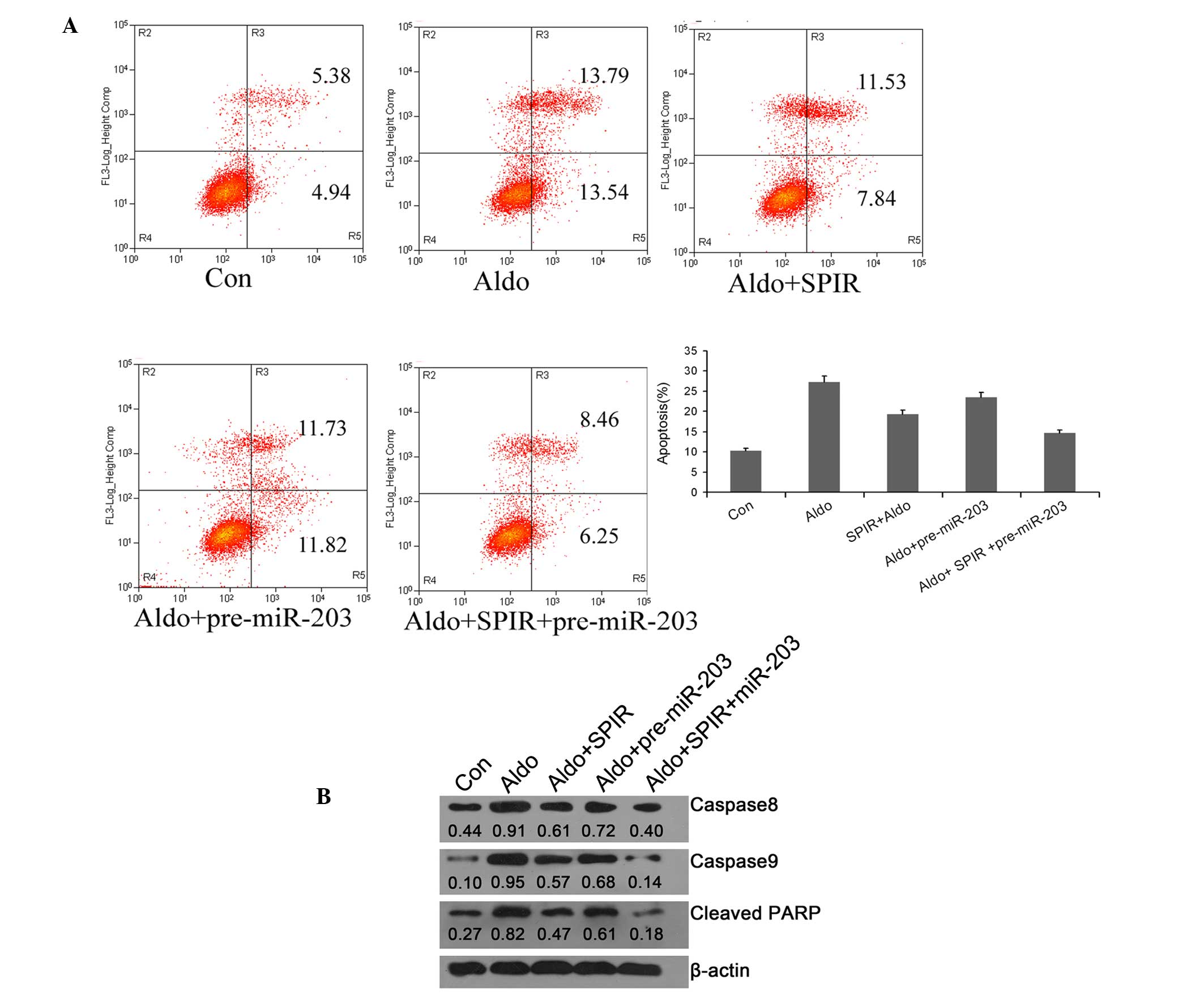
Rno-miR-203 inhibits the apoptosis of
NRK-52E cells treated with Aldo
In order to explore the effect of rno-miR-203 on
cell apoptosis, pre-miR-203 was transfected into NRK-52E cells and
cell apoptosis was analyzed by flow cytometry. Aldo-induced cell
apoptosis was reduced in the cells transfected with pre-miR-203,
suggesting that miR-203 may have a protective role, similar to that
of spironolactone, in preventing cell death induced by high Aldo
concentrations. Notably, when the cells were co-treated with
pre-miR-203 and spironolactone, the cell apoptosis resulting from
exposure to a high Aldo concentration was decreased to a greater
extent as compared with that in the groups treated with pre-miR-203
or spironolactone alone (Fig.
5A).
Rno-miR-203 inhibits apoptosis in
NRK-52E cells treated with Aldo via death receptor and
mitochondrial apoptotic signaling
In order to investigate the mechanism underlying the
rno-miR-203-mediated inhibition of Aldo-induced NRK-52E cell
apoptosis, western blotting was performed to detect the protein
expression levels of the apoptosis initiators, caspase-8 caspase-9
and cleaved PARP. The protein expression levels of caspase-8,
caspase-9 and cleaved PARP were markedly increased in the cells
treated with a high concentration of Aldo. Conversely, when the
cells were transfected with pre-miR-203 or pre-treated with
spironolactone, the protein expression levels of caspase-8,
caspase-9 and cleaved PARP were markedly decreased compared with
those in the Aldo group. In addition, the protein expression levels
of caspase-8, caspase-9 and cleaved PARP were most evidently
decreased in the cells co-treated with pre-miR-203 and
spironolactone (Fig. 5B). These
results suggest that rno-miR-203 may attenuate Aldo-induced cell
apoptosis via the inhibition of death receptor and mitochondrial
apoptotic signaling.
Discussion
AKI is a common condition in patients who are
hospitalized for major surgery or are critically ill, and is
associated with increased mortality (27). Previous reports on the molecular
mechanisms underlying this disease have been conflicting (27,28).
Increasingly, evidence has suggested that apoptosis is the major
mechanism underlying early tubule cell death in contemporary
clinical AKI (29,30). Nevertheless, controversies remain
regarding the contribution of apoptosis to AKI; first, most
estimates place the peak incidence of apoptosis at only 3–5% of
tubule cells following ischemic injury, which is arguably
insufficient to explain the profound renal dysfunction. Second,
apoptosis is more commonly encountered in the distal tubule,
whereas loss of viable cells occurs predominantly in proximal
segments. Third, apoptosis generally is regarded as a physiological
process that removes damaged cells and therefore may be beneficial
to the organ and the organism (29,30).
Therefore, a comprehensive strategy is required in order to fully
delineate the molecular events and pathways underlying the
initiation and progression of AKI, as well as to establish a more
targeted approach in intervention therapies (3).
Renal ischemia is a common cause of AKI in
hospitalized patients. The predominant characteristic of ischemia
is hypoxia (26). Antimycin A is a
mixture of antimycin compounds that can be used as a respiratory
depressant (27). In the present
study, mice models of AKI were established by I/R, and NRK-52E cell
models of AKI by treatment with H/R or antimycin A.
Aldo is an important mediator of the RAAS and has a
pivotal role in the regulation of salt and extracellular fluid
metabolism (28). Furthermore, Aldo
exerts non-hemodynamic effects, leading to end organ damage and
renal injury (31,32). It has previously been reported that
the concentration of Aldo in the renal tissues, urine and plasma is
increased when AKI occurs, and thus it may be used as a marker of
AKI (33). In addition, the Aldo
receptor antagonist, spironolactone, has been shown to exert renal
protective effects; in a previous study, RAAS-induced Kim-1
expression in proximal tubule endothelial cells led to kidney
injury, while treatment with spironolactone reduced Kim-1
expression and attenuated kidney injury (15). These results suggested that Aldo may
be associated with the expression of Kim-1 in renal tissues during
AKI.
The present study performed immunohistochemical
analyses in order to detect the levels of Aldo in the urine of
mice. Furthermore, the expression of Ang II, which is the upstream
mediator of Aldo in the RAAS (34),
was additionally used as a indirect marker of Aldo levels. The
concentration of Aldo was upregulated in the urine of the I/R mice
models and both Ang II and Kim-1 were upregulated in the I/R mice
renal tissues. Conversely, the urine Aldo concentration and Kim-1
expression levels in the renal tissues were significantly reduced
in the mice treated with spironolactone compared with those in the
I/R mice. These results suggest that the upregulation of Kim-1
during AKI was induced by the upregulation of Aldo. Kim-1 was also
upregulated in the H/R or antimycin A-treated cell models. However,
when the cells were pre-treated with spironolactone, the Kim-1
expression levels were decreased significantly. These results
indicated that H/R- or antimycin A-induced renal proximal tubular
cell injury was also mediated by Aldo via the upregulation of Kim-1
expression.
The present study aimed to investigate the molecular
mechanisms underlying the Aldo-induced upregulation of Kim-1 during
AKI, and to determine whether the regulation was the result of
direct or indirect processes. Bioinformatics software predicted
that Kim-1 may be a target gene of rno-miR-203. Therefore, whether
rno-miR-203 is the bridge between Aldo and Kim-1 upregulation in
AKI was investigated in the present study. The expression levels of
rno-miR-203 in the renal tissues of I/R mice and H/R or antimycin
A-treated cells were analyzed by RT-qPCR. The results showed that
the levels of rno-miR-203 were significantly reduced in the renal
tissues of I/R mice compared with those in the controls. In
addition, when I/R mice were fed with spironolactone, the levels of
rno-miR-203 were markedly increased compared with those in the I/R
mice. Similar results were observed in the H/R or antimycin
A-treated cells. Notably, the pattern of expression of rno-miR-203
was the inverse of that of Kim-1, and a direct association between
rno-miR-203 and Kim-1 in NRK-52E cells was verified using
luciferase reporter assays.
Bioinformatics software predicted that there is a
CpG island in the promoter of rno-miR-203. Therefore, it was
hypothesized that hypermethylation may underlie the dysregulation
of rno-miR-203 in AKI, and MSP was performed in order to verify
this hypothesis. Hypermethylated rno-miR-203 was detected in the
renal tissues of I/R mice and the H/R or antimycin A-treated cells;
this suggests that Aldo indirectly induced Kim-1 expression by
mediating the hypermethylation of rno-miR-203. In order to reduce
Kim-1 expression in NRK cells, the cells were pre-treated with
pre-miR-203 and spironolactone prior to treatment with Aldo. Cell
apoptosis was significantly reduced in the spironolactone and
pre-miR-203-treated groups, as compared with the groups treated
with Aldo. Notably, co-treatment with spironolactone and
pre-miR-203 had a greater inhibitory effect on apoptosis, as
compared with either treatment alone.
In conclusion, the present study demonstrated that
Aldo was able to alter the methylation status of rno-miR-203,
promoting its hypermethylation and inducing the expression of
Kim-1. Kim-1 in turn may induce NRK renal cell apoptosis and
necrosis via the death receptor and mitochondrial apoptosis
signaling pathways, leading to AKI. To the best of our knowledge,
this is the first study to report such findings. The results of the
present study suggested that miR-203 may be a promising diagnostic
marker or novel therapeutic target for AKI.
Acknowledgements
The present study was supported by scientific
research funds from the Science and Technology Department of Hunan
Province (grant no. 2012FJ7001) and the Health Department of Hunan
Province (grant no. B2012-012).
References
|
1
|
Dellepiane S, Marengo M and Cantaluppi V:
Detrimental cross-talk between sepsis and acute kidney injury: New
pathogenic mechanisms, early biomarkers and targeted therapies.
Crit Care. 20:612016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Harty J: Prevention and management of
acute kidney injury. Ulster Med J. 83:149–157. 2014.PubMed/NCBI
|
|
3
|
Husi H, Sanchez-Niño MD, Delles C, Mullen
W, Vlahou A, Ortiz A and Mischak H: A combinatorial approach of
proteomics and systems biology in unravelling the mechanisms of
acute kidney injury (AKI): Involvement of NMDA receptor GRIN1 in
murine AKI. BMC Syst Biol. 7:1102013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xu J, Jiang W, Fang Y, Teng J and Ding X:
Management of Cardiac Surgery-Associated Acute Kidney Injury.
Contrib Nephrol. 187:131–142. 2016.PubMed/NCBI
|
|
5
|
Ftouh S and Lewington A: Acute Kidney
Injury Guideline Development Group convened by the National
Clinical Guidelines Centre and commissioned by the National
Institute for Health and Care Excellence, in association with The
Royal College of Physicians' Clinic: Prevention, detection and
management of acute kidney injury: Concise guideline. Clin Med.
14:61–65. 2014. View Article : Google Scholar
|
|
6
|
Gaffney AM and Sladen RN: Acute kidney
injury in cardiac surgery. Curr Opin Anaesthesiol. 28:50–59. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McKay DB: Intracellular pattern
recognition receptors and renal ischemia. Crit Rev Immunol.
31:297–306. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huang CH, Lai CC, Yang AH and Chiang SC:
Myocardial preconditioning reduces kidney injury and apoptosis
induced by myocardial ischaemia and reperfusion. Eur J Cardiothorac
Surg. 48:382–391. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Prozialeck WC, Edwards JR, Lamar PC, Liu
J, Vaidya VS and Bonventre JV: Expression of kidney injury
molecule-1 (Kim-1) in relation to necrosis and apoptosis during the
early stages of Cd-induced proximal tubule injury. Toxicol Appl
Pharmacol. 238:306–314. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Waanders F, Vaidya VS, van Goor H,
Leuvenink H, Damman K, Hamming I, Bonventre JV, Vogt L and Navis G:
Effect of renin-angiotensin-aldosterone system inhibition, dietary
sodium restriction, and/or diuretics on urinary kidney injury
molecule 1 excretion in nondiabetic proteinuric kidney disease: A
post hoc analysis of a randomized controlled trial. Am J Kidney
Dis. 53:16–25. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Waanders F, van Timmeren MM, Stegeman CA,
Bakker SJ and van Goor H: Kidney injury molecule-1 in renal
disease. J Pathol. 220:7–16. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bonventre JV: Kidney injury molecule-1
(KIM-1): A urinary biomarker and much more. Nephrol Dial
Transplant. 24:3265–3268. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Han WK, Waikar SS, Johnson A, Betensky RA,
Dent CL, Devarajan P and Bonventre JV: Urinary biomarkers in the
early diagnosis of acute kidney injury. Kidney Int. 73:863–869.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen C, Liang W, Jia J, van Goor H,
Singhal PC and Ding G: Aldosterone induces apoptosis in rat
podocytes: Role of PI3-K/Akt and p38MAPK signaling pathways.
Nephron Exp Nephrol. 113:e26–e34. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
de Borst MH, van Timmeren MM, Vaidya VS,
de Boer RA, van Dalen MB, Kramer AB, Schuurs TA, Bonventre JV,
Navis G and van Goor H: Induction of kidney injury molecule-1 in
homozygous Ren2 rats is attenuated by blockade of the
renin-angiotensin system or p38 MAP kinase. Am J Physiol Renal
Physiol. 292:F313–F320. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vrba L, Jensen TJ, Garbe JC, Heimark RL,
Cress AE, Dickinson S, Stampfer MR and Futscher BW: Role for DNA
methylation in the regulation of miR-200c and miR-141 expression in
normal and cancer cells. PloS One. 5:e86972010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kusenda B, Mraz M, Mayer J and Pospisilova
S: Re. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub.
150:205–215. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Aguado-Fraile E, Ramos E, Conde E,
Rodríguez M, Liaño F and García-Bermejo ML: MicroRNAs in the
kidney: Novel biomarkers of acute kidney injury. Nefrologia.
33:826–834. 2013.PubMed/NCBI
|
|
19
|
Lorenzen JM, Kielstein JT, Hafer C, Gupta
SK, Kümpers P, Faulhaber-Walter R, Haller H, Fliser D and Thum T:
Circulating miR-210 predicts survival in critically ill patients
with acute kidney injury. Clin J Am Soc Nephrol. 6:1540–1546. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Du J, Cao X, Zou L, Chen Y, Guo J, Chen Z,
Hu S and Zheng Z: MicroRNA-21 and risk of severe acute kidney
injury and poor outcomes after adult cardiac surgery. PloS One.
8:e633902013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lan YF, Chen HH, Lai PF, Cheng CF, Huang
YT, Lee YC, Chen TW and Lin H: MicroRNA-494 reduces ATF3 expression
and promotes AKI. J Am Soc Nephrol. 23:2012–2023. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bellinger MA, Bean JS, Rader MA,
Heinz-Taheny KM, Nunes JS, Haas JV, Michael LF and Rekhter MD:
Concordant changes of plasma and kidney microRNA in the early
stages of acute kidney injury: Time course in a mouse model of
bilateral renal ischemia-reperfusion. PloS One. 9:e932972014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wilflingseder J, Sunzenauer J, Toronyi E,
Heinzel A, Kainz A, Mayer B, Perco P, Telkes G, Langer RM and
Oberbauer R: Molecular pathogenesis of post-transplant acute kidney
injury: Assessment of whole-genome mRNA and miRNA profiles. PloS
One. 9:e1041642014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zager RA, Johnson AC, Hanson SY and Lund
S: Ischemic proximal tubular injury primes mice to
endotoxin-induced TNF-alpha generation and systemic release. Am J
Physiol Renal Physiol. 289:F289–F297. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tokumoto M, Lee JY, Fujiwara Y and Satoh
M: Alteration of DNA binding activity of transcription factors in
NRK-52E rat proximal tubular cells treated with cadmium. J Toxicol
Sci. 39:735–738. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Arocho A, Chen B, Ladanyi M and Pan Q:
Validation of the 2-DeltaDeltaCt calculation as an alternate method
of data analysis for quantitative PCR of BCR-ABL P210 transcripts.
Diagn Mol Pathol. 15:56–61. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Husi H and Human C: Molecular determinants
of acute kidney injury. J Inj Violence Res. 7:75–86.
2015.PubMed/NCBI
|
|
28
|
Devarajan P: Update on mechanisms of
ischemic acute kidney injury. J Am Soc Nephrol. 17:1503–1520. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dagher PC: Apoptosis in ischemic renal
injury: Roles of GTP depletion and p53. Kidney Int. 66:506–509.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kaushal GP, Basnakian AG and Shah SV:
Apoptotic pathways in ischemic acute renal failure. Kidney Int.
66:500–506. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
López-Andrés N, Iñigo C, Gallego I, Diez J
and Fortuño MA: Aldosterone induces cardiotrophin-1 expression in
HL-1 adult cardiomyocytes. Endocrinology. 149:4970–4978. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
White LE, Hassoun HT, Bihorac A, Moore LJ,
Sailors RM, McKinley BA, Valdivia A and Moore FA: Acute kidney
injury is surprisingly common and a powerful predictor of mortality
in surgical sepsis. J Trauma Acute Care Surg. 75:432–438. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sawada H, Naito Y, Oboshi M, Iwasaku T,
Okuhara Y, Morisawa D, Eguchi A, Hirotani S and Masuyama T: Iron
restriction inhibits renal injury in aldosterone/salt-induced
hypertensive mice. Hypertens Res. 38:317–322. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chun TY, Chander PN, Kim JW, Pratt JH and
Stier CT Jr: Aldosterone, but not angiotensin II, increases
profibrotic factors in kidney of adrenalectomized stroke-prone
spontaneously hypertensive rats. Am J Physiol Endocrinol Metab.
295:E305–E312. 2008. View Article : Google Scholar : PubMed/NCBI
|