Introduction
Neuronal injury during status epilepticus (SE) is
the result of increased excitotoxicity caused by calcium entrance
into the cells leading to activation of various protease-mediated
cascades and mitochondrial and nucleic injury, resulting in cell
death (1). Research have indicated
that apoptosis plays a critical role in neuronal death following SE
(2), in which caspase-3 is the final
effector in the apoptotic signaling pathway (3,4). In
addition, other studies have demonstrated that the antiapoptotic
protein B-cell lymphoma 2 (Bcl-2) protects against seizure-induced
brain damage (5,6). It is hypothesized that Bcl-2 inhibits
apoptosis by acting on the caspase-3 dependent proteolytic cascade,
and may attenuate SE-induced neuronal damage at least partially via
the suppression of caspase-3 activity (7,8).
MiRs are short non-coding RNAs that regulate gene
expression at the post-transcription and/or translation levels
(9,10). MiR-497 is among the most prominently
downregulated miRs in stroke-induced neuronal death and
neurodegenerative disease (11,12).
Evidence suggests that miR-497 expression is inversely correlated
with the regulation of apoptosis (13,14), and
has been shown to directly target Bcl-2 mRNA (14,15).
Additionally, a few groups have shown the involvement of miRs in
the pathogenesis of epilepsy (16–18).
Baicalin is a flavonoid compound isolated from
Scutellaria baicalensis Georgi, a traditional Chinese
medicinal herb that is speculated to possesses anti-oxidative
(19), anti-inflammatory (20), antiapoptotic (21) and anti-cancer properties (22). For example, baicalin has been shown
to have a protective effect on permanent cerebral ischemia in rats
through anti-inflammatory and antiapoptotic properties, possibly
through the downregulation of nitric oxide synthase and
cyclooxygenase-2 mRNA and cleaved caspase-3 protein expression
(23). While a previous study showed
that baicalin attenuates hippocampal injury after SE in mice
(24), little is known about the
mechanisms underlying this neuroprotective effect.
The present study investigated the role of cerebral
miR-497 as a factor in the regulation of seizure-induced neuronal
death after SE in mice. In addition, the study aimed to determine
whether the neuroprotective effect of baicalin in this model may be
associated with the downregulated expression of miR-497 and
upregulation of the expression of a known, antiapoptotic target
gene, Bcl-2, which results in reduced caspase-3 cleavage.
Materials and methods
Experimental animals
A total of 99 adult ICR male mice weighing 22–28 g
(Shanghai Laboratory Animal Center, Chinese Academy of Sciences,
Shanghai, China) were used in this study. All animals were housed
using an alternating 12-h light/dark cycle in a temperature
(18–25°C) and humidity (50–60%) controlled environment. Food and
water were available ad libitum. The mice were allowed to
adapt to laboratory conditions for at least 1 week before starting
the experiment. Mice were divided into three groups as follows: i)
Sham control group (n=33), which underwent sham operation and
received vehicle (Beijing Zhongshan Golden Bridge Biotechnology
Co., Ltd.); ii) SE group (n=33), which was subjected to kainic acid
(KA) injection and received vehicle; and iii) baicalin group
(n=33), which was subjected to KA injection and treated with
baicalin 100 mg/kg. This study was approved by the Institutional
Animal Care, Ethics and Use Committees of Fujian Medical University
(Fuzhou, China).
Seizure induction
All surgical procedures were carried out in
accordance with the National Institute of Health Guide for the Care
and Use of Laboratory Animals and performed using sterile/aseptic
techniques in accordance with institutional guidelines. Focal-onset
SE was induced by intracerebral ventricle stereotaxic
microinjection of KA (Sigma-Aldrich, St. Louis, MO, USA), and was
performed as previously described (25). Briefly, mice were anesthetized with
an intraperitoneal injection of chloral hydrate (300 mg/kg;
Sigma-Aldrich) and placed in a stereotaxic frame (Gilson, Inc.,
Villiers le Bel, France). Following a midline scalp incision, the
bregma was located. The skull was then drilled for placement of a
guide cannula (bregma coordinates; AP=1.9 mm, L=2.1 mm) based on a
mouse brain stereotaxic atlas (26).
Then, the animal was restrained while an injection cannula was
lowered 2.4 mm below the brain surface into the lateral ventricle
for injection of KA. The injected dose was 0.8 µg KA in 5 µl normal
saline at 1 µl/min, to elicit a prolonged SE. Non-seizure control
mice underwent the same surgical procedures but received
intracerebral ventricle vehicle (normal saline, 5 µl). During the
surgery, rectal temperature was controlled at 37°C using a heating
pad and heat lamp (Beijing Cinontech Co., Ltd., Beijing, China;
http://www.ysf2005.bioon.com.cn/product_606.html).
All efforts were made to minimize the number of animals used and to
avoid pain and suffering. In all cases, Racine stage 5 (rearing and
falling) seizures were observed within 1 h following KA
administration (27).
Drug administration
Baicalin (purity, >95%; CAS Number: 21967-41-9;
Sigma-Aldrich) was dissolved in normal saline and injected
intraperitoneally into mice assigned to the baicalin group at 1 and
8 h after the onset of SE. Mice in the SE group were injected with
normal saline at the same volume and time points as the baicalin
group.
Preparation of brain tissue slices for
hematoxylin and eosin (HE) staining
A total of 18 mice were selected for HE staining
(Beyotime Institute of Biotechnology, Shanghai, China) (6 per
group), and were anesthetized using chloral hydrate (10%, 3.5
ml/kg, i.p.), then sacrificed 72 h after the onset of SE using a
transcardiac infusion of normal saline followed by 4%
paraformaldehyde to fix the brain. The mice were then decapitated
and the fixed brains were removed and embedded in paraffin. The
brain was then cut into 5–6 mm thick sections containing the
hippocampus, which were further cut with a HM340E microtome (Microm
International GmbH, Walldorf, Germany) into 30 µm thick coronal
sections. Then, the sections were deparaffinized with xylene
(Guangzhou Chemical Reagent Factory, Guangzhou, China) and
rehydrated with graded alcohol. Subsequently, the slices were
stained using HE, mounted to glass slides and coverslipped using
neutral gum (ZLI-9516; Zhongshan Golden Bridge Biotechnology Co.,
Ltd.).
Terminal transferase-mediated dUTP
nick end-labeling (TUNEL) analysis
TUNEL staining was carried out using the in situ
cell death detection kit (Promega Corporation, Fitchburg, WI, USA)
according to the manufacturer's instructions. Briefly, 18 mice (6
per group) were anesthetized, then sacrificed at 72 h following
onset of SE by transcardiac perfusion of normal saline followed by
4% paraformaldehyde. The animals were then decapitated with brain
tissue removed and processed as shown above. However, in addition
to the aforementioned procedure, after deparaffinizing the sections
were digested using proteinase K (Zhongshan Golden Bridge
Biotechnology Co., Ltd.). The slides were placed in the
equilibration buffer and then incubated in TdT enzyme (Promega
Corporation) at 37°C for 60 min, followed by 2xSSC (Zhongshan
Golden Bridge Biotechnology Co., Ltd.). to stop the reaction.
Endogenous peroxidase activity was then blocked with 0.3%
H2O2 and the sections were incubated in a
streptavidin horseradish-peroxidase solution (Zhongshan Golden
Bridge Biotechnology Co., Ltd.) for 30 min at room temperature.
3,3′-Diaminobenzidine (Zhongshan Golden Bridge Biotechnology Co.,
Ltd.) was used as chromogen and the sections were counterstained
with hematoxylin (Zhongshan Golden Bridge Biotechnology Co., Ltd.).
The sections were then mounted on glass slides and coverslipped
using neutral gum (ZLI-9516; Zhongshan Golden Bridge Biotechnology
Co., Ltd.). The TUNEL-positive cells and total cells in the
hippocampal CA3 subfields were manually counted while viewed at
×400 magnification using a microscope (Olympus Corporation, Tokyo,
Japan). The number of TUNEL-positive cells was expressed as the
percentage of total counted cells.
Immunohistochemistry
A total of 18 mice (6 per group) were anesthetized
72 h following SE induction then sacrificed by transcardiac
perfusion of normal saline followed by 4% paraformaldehyde.
Decapitation was performed, with the brains removed and processed
as shown above. However, the paraffin-embedded sections were
deparaffinized with xylene and rehydrated graded alcohol, followed
by treatment with 1% hydrogen peroxide to eliminate endogenous
peroxidase activity. After treatment with 5% goat serum reagent
(Zhongshan Golden Bridge Biotechnology Co., Ltd.) at room
temperature for 30 min, the sections were then incubated overnight
at 4°C with a rabbit polyclonal anti-Bcl-2 antibody (1:50; ZA-0536;
Zhongshan Golden Bridge Biotechnology Co., Ltd.). The sections were
incubated with biotinylated goat anti-rabbit secondary antibody
(1:200; ZDR-5403; KPL, Inc., Zhongshan Golden Bridge Biotechnology
Co., Ltd.) at room temperature for 60 min, followed by incubation
with a streptavidin-biotin peroxidase complex solution (Wuhan
Boster Biological Technology, Ltd., Wuhan, China) at room
temperature for 120 min. Subsequently, the sections were stained
using 3,3′-diaminobenzidine, counterstained with hematoxylin,
dehydrated, then mounted and coverslipped using neutral gum
(ZLI-9516; Zhongshan Golden Bridge Biotechnology Co., Ltd.). All
sections were processed with Image-Pro Plus (Media Cybernetics,
Inc., Rockville, MD, USA) for imaging and analysis.
Western blot analysis
For western blot analysis, 18 mice (6 per group)
were anesthetized at 72 h following the induction of SE, sacrificed
via transcardiac perfusion of normal saline followed by 4%
paraformaldehyde, then decapitated for brain removal. The
hippocampus was dissected, homogenized and purified using protein
extraction reagents according to the manufacturer's instructions
(KeyGen Biotech, Co., Ltd., Nanjing, China). Protein concentration
was determined by the Bradford method using bovine serum albumin
(Beyotime Institute of Biotechnology) as the standard. Different
samples with an equal quantity of protein (20 mg) were separated on
10% SDS-polyacrylamide gels (Wuhan Boster Biological Technology,
Ltd.), transferred to nitrocellulose membranes (Membrane Solutions
Co., Ltd., Shanghai, China), and blocked in 5% nonfat dry milk
buffer (Wuhan Boster Biological Technology, Ltd.). β-Actin (Cell
Signaling Technology, Inc., Danvers, MA, USA) was used as a loading
control. The membranes were then incubated overnight at 4°C with a
rabbit polyclonal anti-Bcl-2 antibody (1:200; ZA-0536; Zhongshan
Golden Bridge Biotechnology Co., Ltd.,) and cleaved caspase-3
(1:800; Cell Signaling Technology, Inc.), followed by incubation
with horseradish-peroxidase conjugated secondary antibodies
(1:2,000; SC-2004; Zhongshan Golden Bridge Biotechnology Co.,
Ltd.). Protein expression was detected using an enhanced
chemiluminescence detection system (20-500-120; Bio-Rad
Laboratories, Inc., Hercules, CA, USA) and exposed on X-ray film
(Bio-Rad Laboratories, Inc.). The optical densities of Bcl-2 (26
kDa), cleaved caspase-3 (17 kDa) and β-actin (42 kDa) bands on the
X-ray film were quantitatively analyzed using Quantity One software
version 4.6.1 (Bio-Rad Laboratories). The results were expressed as
the ratios of Bcl-2 and cleaved caspase-3 to β-actin.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
For RT-qPCR, 27 mice (9 per group) were
anesthetized, then sacrificed via decapitation 12 h following
induction of SE. The brain was carefully removed, and the
hippocampus dissected. Total RNA was extracted using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc., Carlsbad, CA, USA),
according to the manufacturer's protocol. No additional DNase
treatment was conducted after the RNA was extracted from the
sample. Next, 2 µg total RNA underwent reverse transcription using
an miR-497-specific RT-primer along with a Bulge-Loop™ miRNA
qRT-PCR Primer Set (RiboBio Co., Ltd., Guangzhou, China), according
to the manufacturer's instructions. Hot start Taq polymerase as
well as the forward and reverse primers for all of the genes were
included in the Bulge-Loop™ miRNAqRT-PCR starter kit (RiboBio Co.,
Ltd.). The reaction mixture included 3.6 µl RNase-free
H2O, 0.2 µl forward and reverse primers, 5 µl SYBR Green
supermix and 1 µl cDNA. SYBR Green I stain is maximally excited at
497 nm, but also has secondary excitation peaks at ~290 nm and 380
nm. The fluorescence emission of SYBR Green I stain bound to DNA is
centered at 520 nm. PCR was conducted using a CFX-96 PCR machine
(Bio-Rad Laboratories, Inc., Hercules, CA, USA), and the following
conditions were used: Pre-denaturation at 95°C for 3 min, 40 cycles
of 95°C for 10 sec, 60°C for 20 sec and 70°C for 10 sec.
The ubiquitously expressed U6 small nuclear RNA was
used for normalization. All primers used for qPCR were included in
the commercial kit. Each sample was run in triplicate and qPCR data
were analyzed using the 2−ΔΔCq method (28) and the software used to analyze the
data was called BioRAD CFX Manager 2.1 (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Statistical analysis
Statistical analyses were performed using SPSS
software, version 18.0 (SPSS, Inc., Chicago, IL, USA). Quantitative
data were represented as mean ± standard deviation. Differences
among groups were compared using one-way analysis of variance with
a Least Significant Difference post-hoc pair-wise
comparison. For the apoptosis data, as the sham group had a value
of zero for each sample, a one-sample t-test was applied for SE and
baicalin groups to test the mean=0, which indicates a comparison
with the sham group. Furthermore, a two-sample t-test was used to
compare the difference between SE and baicalin groups. All the
statistical assessments were two tailed and P<0.05 was
considered as signifying a significant difference.
Results
SE
The first signs of seizures occurred within 1 h
after KA administration and continued for an additional 1–3 h.
During seizures, mice displayed manifestations common to limbic SE
as previously characterized by Racine et al (27) as stage I–V, including stereotyped
masticatory movements (stage I), head movements (stage II),
forelimb clonus (stage III), rearing (stage IV) and rearing and
falling (stage V).
Morphological examination
Morphological examination was performed on sections
cut from brains removed 72 h following SE onset, fixed with 4%
paraformaldehyde and stained using HE. As shown in Fig. 1, the majority of neurons in the
hippocampal CA1 and CA3 subfields of mice in the SE group appeared
shrunken with eosinophilic cytoplasm, triangulated pyknotic nuclei
and cavitation (empty holes indicative of vapor cavities; Fig. 1B and E). In the sham control group,
there was no evidence of neuronal damage, with a lack of
eosinophilic cytoplasm or triangulated pyknotic nuclei evident in
the brains of mice in the SE group (Fig.
1A and D). Additionally, while mice that received baicalin (100
mg/kg) prior to SE showed eosinophilic cytoplasm and triangulated
pyknotic nuclei indicative of SE induced neuronal damage, the
number of damaged cells in the same areas was reduced compared to
SE mice who did not receive baicalin (Fig. 1C and F).
Baicalin inhibits neuronal apoptosis
after SE
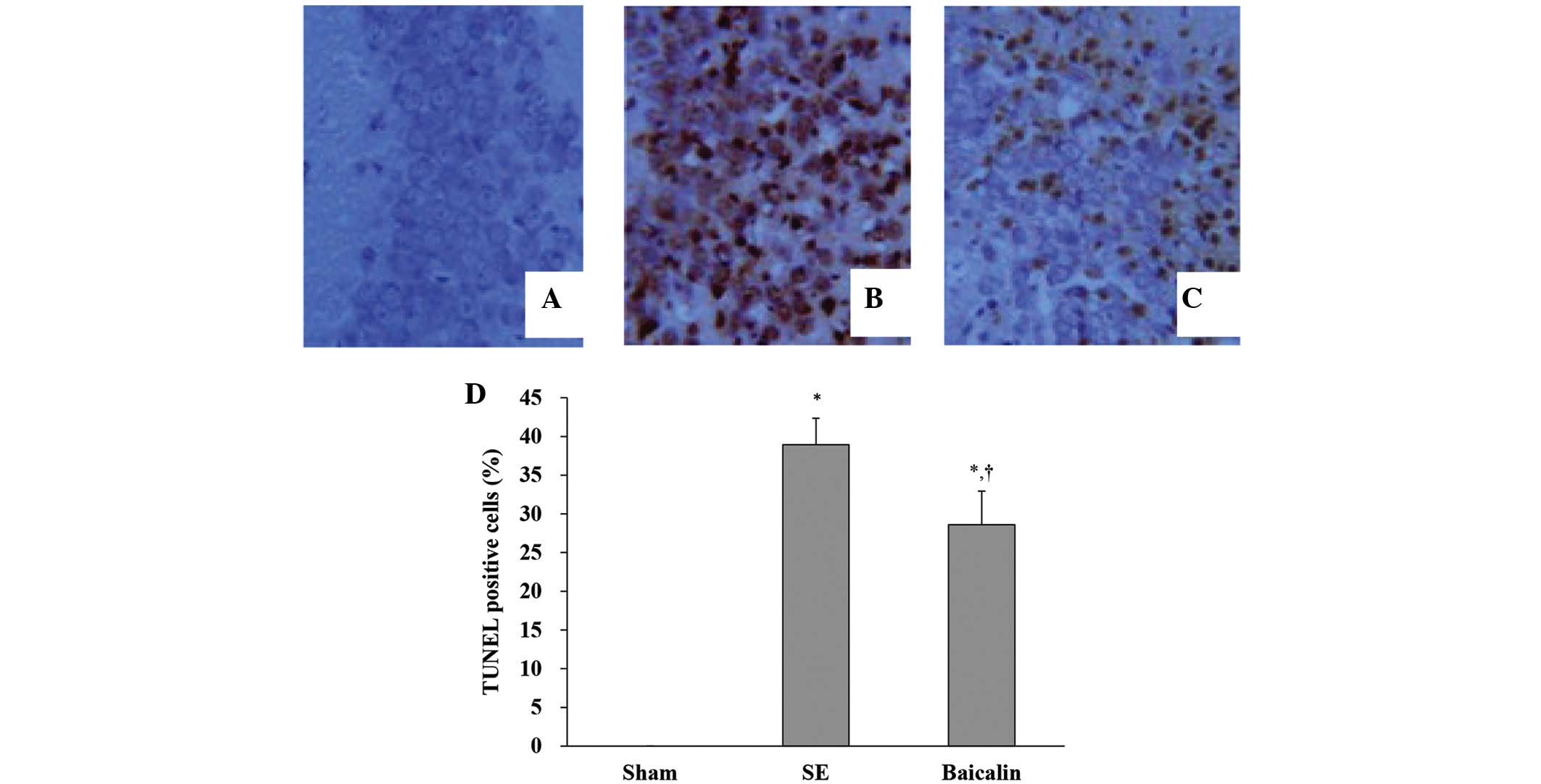
TUNEL staining was used to evaluate apoptotic
neuronal death in the hippocampal CA3 subfields of mice sacrificed
72 h following the induction of SE, processed as described,
digested using proteinase K with chromosomal degradation visualized
and using TdT followed by chromogen. Fig. 2 shows representative images of
neuronal apoptosis in hippocampal CA3 subfields for each treatment
group. Quantitative analysis of apoptotic nuclei as expressed as
the percentage of apoptotic cells of the total revealed that the SE
and baicalin groups had higher levels of TUNEL staining indicative
of neuronal apoptosis compared with the sham group. By contrast,
those that received baicalin treatment prior to SE had
significantly lower levels of TUNEL staining than those that did
not, indicating a suppression of SE-induced neuronal apoptosis
(Fig. 2D) (P<0.05).
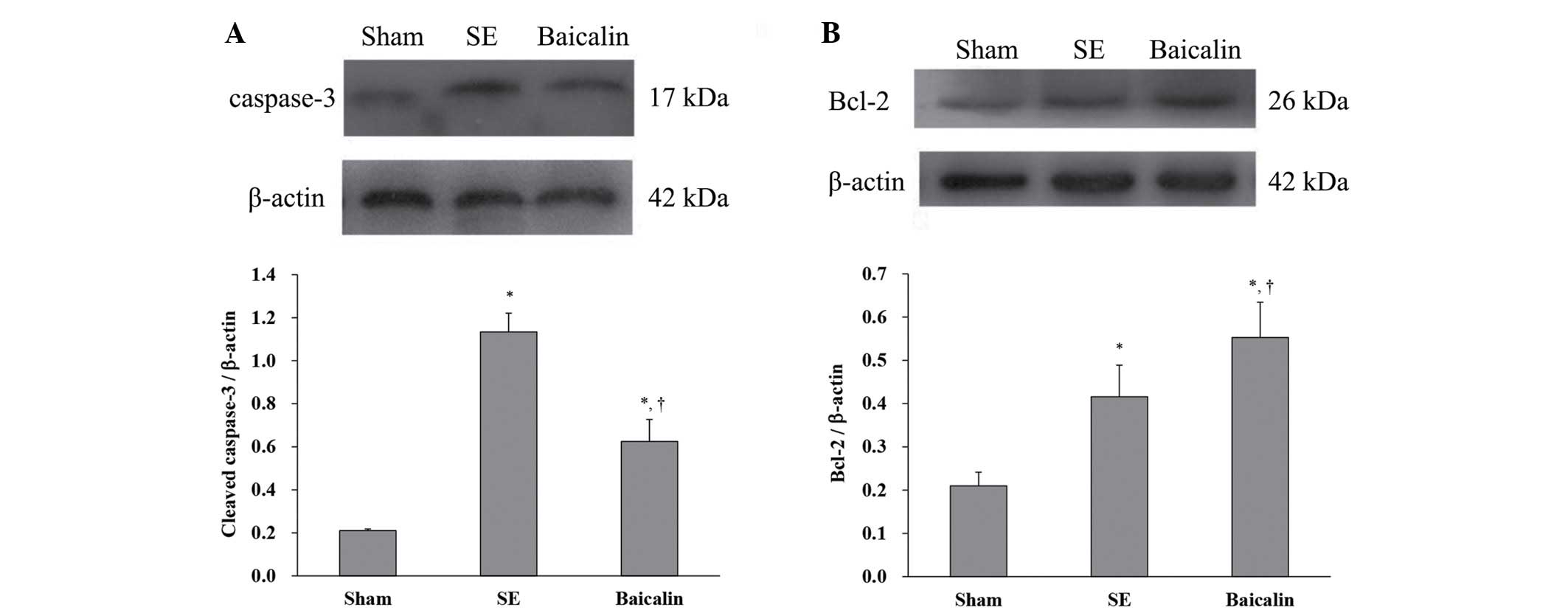
The levels of cleaved caspase-3 and Bcl-2 (relative
to β-actin) in brain tissue removed from mice sacrificed 72 h
following the induction of SE was analyzed using western blotting.
The results showed that SE increased cleaved caspase-3 protein
expression, which was suppressed by baicalin (Fig. 3A) (P<0.05). By contrast, SE
upregulated antiapoptotic Bcl-2 protein expression (SE, 0.42±0.07
vs. sham, 0.21±0.03; P<0.001) (Fig.
3B). Baicalin treatment resulted in greater increases in Bcl-2
expression as a result of SE compared with SE alone (baicalin,
0.55±0.08 vs. SE, 0.42±0.07; P=0.003) (Fig. 3B). These results suggest that
baicalin administration following KA-induced SE leads to lower
expression of proteins associated with neuronal apoptosis in the
hippocampus.
Expression of Bcl-2 within the hippocampal CA1 and
CA3 subfields of brain sections from mice sacrificed 72 h after the
induction of SE was analyzed by immunohistochemistry using
antibodies raised against Bcl-2 and visualized using horseradish
peroxidase/biotin labeling. Representative hippocampal images of
Bcl-2 staining from the three treatment groups are shown in
Fig. 4. Quantification of Bcl-2
positive cells in the CA1 (Fig. 5A)
and CA3 (Fig. 5B) subfields showed
that SE increased the number of Bcl-2 positive cells, which were
further enhanced by baicalin treatment (CA1: baicalin, 22.79±4.19
vs. SE, 14.67±2.89; P=0.001) (CA3: baicalin, 39.34±4.67 vs. SE,
23.90±5.47; P<0.001).
Baicalin inhibits the expression of
miR-497
Expression of mature miR-497 was analyzed using
RT-qPCR, within the CA3 and CA1 subfields of the hippocampus from
mice sacrificed at 12 h after SE via decapitation and compared with
samples from time-matched, vehicle-injected, non-seizure controls.
The results showed that in the hippocampus of SE mice, expression
of miR-497 was increased compared with the sham (SE, 1.62±0.32 vs.
sham, 1.03±0.15; P<0.001). However, baicalin treatment following
SE reduced miR-497 expression as compared to SE alone (baicalin,
1.33±0.25 vs. SE, 1.62±0.32, P=0.02; Fig. 6A). Thus, as shown in Fig. 6B, SE may induce elevation of miR-497
levels, which may negatively regulate expression of Bcl-2.
Discussion
In the present study, intracerebral ventricle
microinjection of KA was used to trigger SE, leading to the
emergence of epileptic seizures and subsequent hippocampal damage.
Previous studies have confirmed that apoptotic pathways contribute
to seizure-induced neuronal death in this model (4,5,24,29), as
evidenced by significantly altered hippocampal damage in animals
lacking apoptosis-associated genes and proteins, including miR-497,
Bcl-2 and caspase-3. To the best of our knowledge, the present
study is the first to show that baicalin not only significantly
reduces the number of TUNEL-positive cells following SE, but also
alters the expression of Bcl-2 and cleaved caspase-3 protein, in
addition to miR-497. However, while it may be hypothesized that
baicalin exerts its neuroprotective effects via the downregulation
of miR-497, leading to the upregulation of the caspase-3 inhibitor
Bcl-2, the precise mechanisms underlying the action of baicalin
remain poorly understood and will require further study.
The present finding that SE increases Bcl-2
expression in the hippocampal CA1 and CA3 regions is consistent
with a previous report (30). It was
also found that baicalin significantly enhanced the number of cells
expressing Bcl-2, while reducing miR-497 expression and caspase-3
protein activation. To our knowledge, this is the first
demonstration of the efficacy of baicalin in enhancing the effect
of SE on Bcl-2 expression while reducing caspase-3 cleavage.
In this study, we reported that miR-497 is rapidly
upregulated in the hippocampus following KA induced SE in mice,
demonstrating that miR-497 may be associated with early
pathological changes and seizure-induced neuronal death. As miR-497
is known to downregulate the expression of Bcl-2 (14,15),
SE-induced expression of miR-497 may promote apoptosis by limiting
post SE increases in Bcl-2 levels, as Bcl-2 has a well-established
inhibitory effect on caspase-3 activation (8). However, it was presently found that
baicalin inhibited SE-induced increases in miR-497 expression and
enhanced SE-induced increases in Bcl-2 protein levels. Therefore,
we hypothesize that the suppression of caspase-3 activation by
baicalin is mediated by the inhibition of the expression of
miR-497, subsequently disinhibiting the expression of the caspase-3
inhibitor Bcl-2.
In conclusion, the present study demonstrated that
baicalin exerts a neuroprotective effect against apoptotic damage
as a result of KA-induced epileptic seizures. This study presents
further support to the potential use of baicalin as a therapeutic
agent to reduce seizure-induced brain injury. We hypothesize that
baicalin may limit delayed neuronal death by preventing miR-497
from inhibiting Bcl-2 expression, which leads to decreased levels
of cleaved caspase-3 protein and subsequent apoptosis in epileptic
insults. While it has previously been shown that miR-497 regulates
neuronal death following ischemia (23), the present study is the first to
identify an association between miR497 and seizure-induced damage.
However, additional studies will be necessary to determine the
precise mechanisms involved in the impact of baicalin on
seizure-induced neuronal death. Furthermore, additional studies are
required to determine effect of baicalin on normal controls,
thereby determining if its effect is non-specific or directly
related to SE. This study further demonstrates the role of miRs in
regulating the antiapoptotic Bcl-2 family proteins and proapoptotic
caspase family proteins.
Acknowledgements
This study was supported by the Natural Science
Foundation of Fujian Province, China (grant no. 2011J01175), and by
internal funding from the Department of Neurosurgery, Union
Hospital of Fujian Medical University.
Glossary
Abbreviations
Abbreviations:
|
HE
|
hematoxylin and eosin
|
|
KA
|
kainic acid
|
|
SE
|
status epilepticus
|
References
|
1
|
Henshall DC and Simon RP: Epilepsy and
apoptosis pathways. J Cereb Blood Flow Metab. 25:1557–1572. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lopez-Meraz ML, Niquet J and Wasterlain
CG: Distinct caspase pathways mediate necrosis and apoptosis in
subpopulations of hippocampal neurons after status epilepticus.
Epilepsia. 51(Suppl 3): S56–S60. 2010. View Article : Google Scholar
|
|
3
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kondratyev A and Gale K: Intracerebral
injection of caspase-3 inhibitor prevents neuronal apoptosis after
kainic acid-evoked status epilepticus. Brain Res Mol Brain Res.
75:216–224. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Henshall DC, Araki T, Schindler CK, Lan
JQ, Tiekoter KL, Taki W and Simon RP: Activation of
Bcl-2-associated death protein and counter-response of Akt within
cell populations during seizure-induced neuronal death. J Neurosci.
22:8458–8465. 2002.PubMed/NCBI
|
|
6
|
Uysal H, Cevik IU, Soylemezoglu F, Elibol
B, Ozdemir YG, Evrenkaya T, Saygi S and Dalkara T: Is the cell
death in mesial temporal sclerosis apoptotic? Epilepsia.
44:778–784. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hua F, Cornejo MG, Cardone MH, Stokes CL
and Lauffenburger DA: Effects of Bcl-2 levels on Fas
signaling-induced caspase-3 activation: Molecular genetic tests of
computational model predictions. J Immunol. 175:985–995. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Swanton E, Savory P, Cosulich S, Clarke P
and Woodman P: Bcl-2 regulates a caspase-3/caspase-2 apoptotic
cascade in cytosolic extracts. Oncogene. 18:1781–1787. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ambros V: microRNAs: Tiny regulators with
great potential. Cell. 107:823–826. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Barak B, Shvarts-Serebro I, Modai S, Gilam
A, Okun E, Michaelson DM, Mattson MP, Shomron N and Ashery U:
Opposing actions of environmental enrichment and Alzheimer's
disease on the expression of hippocampal microRNAs in mouse models.
Transl Psychiatry. 3:e3042013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jeyaseelan K, Lim KY and Armugam A:
MicroRNA expression in the blood and brain of rats subjected to
transient focal ischemia by middle cerebral artery occlusion.
Stroke. 39:959–966. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yadav S, Pandey A, Shukla A, Talwelkar SS,
Kumar A, Pant AB and Parmar D: miR-497 and miR-302b regulate
ethanol-induced neuronal cell death through BCL2 protein and cyclin
D2. J Biol Chem. 286:37347–37357. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yin KJ, Deng Z, Huang H, Hamblin M, Xie C,
Zhang J and Chen YE: miR-497 regulates neuronal death in mouse
brain after transient focal cerebral ischemia. Neurobiol Dis.
38:17–26. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhu W, Zhu D, Lu S, Wang T, Wang J, Jiang
B, Shu Y and Liu P: miR-497 modulates multidrug resistance of human
cancer cell lines by targeting BCL2. Med Oncol. 29:384–391. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hu K, Zhang C, Long L, Long X, Feng L, Li
Y and Xiao B: Expression profile of microRNAs in rat hippocampus
following lithium-pilocarpine-induced status epilepticus. Neurosci
Lett. 488:252–257. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kan AA, van Erp S, Derijck AA, de Wit M,
Hessel EV, O'Duibhir E, de Jager W, Van Rijen PC, Gosselaar PH, de
Graan PN and Pasterkamp RJ: Genome-wide microRNA profiling of human
temporal lobe epilepsy identifies modulators of the immune
response. Cell Mol Life Sci. 69:3127–3145. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pitkänen A and Lukasiuk K: Molecular
biomarkers of epileptogenesis. Biomark Med. 5:629–633. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cheng F, Lu Y, Zhong X, Song W, Wang X,
Sun X, Qin J, Guo S and Wang Q: Baicalin's therapeutic time window
of neuroprotection during transient focal cerebral ischemia and its
antioxidative effects in vitro and in vivo. Evid Based Complement
Alternat Med. 2013:1202612013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li L, Bao H, Wu J, Duan X, Liu B, Sun J,
Gong W, Lv Y, Zhang H, Luo Q, et al: Baicalin is anti-inflammatory
in cigarette smoke-induced inflammatory models in vivo and in
vitro: A possible role for HDAC2 activity. Int Immunopharmacol.
13:15–22. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cao Y, Mao X, Sun C, Zheng P, Gao J, Wang
X, Min D, Sun H, Xie N and Cai J: Baicalin attenuates global
cerebral ischemia/reperfusion injury in gerbils via anti-oxidative
and anti-apoptotic pathways. Brain Res Bull. 85:396–402. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen J, Li Z, Chen AY, Ye X, Luo H, Rankin
GO and Chen YC: Inhibitory effect of baicalin and baicalein on
ovarian cancer cells. Int J Mol Sci. 14:6012–6025. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tu XK, Yang WZ, Shi SS, Wang CH and Chen
CM: Neuroprotective effect of baicalin in a rat model of permanent
focal cerebral ischemia. Neurochem Res. 34:1626–1634. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ouyang LQ, Liang RS, Yang WZ, Chen CM, Tu
XK and Wen S: Neuroprotective effect of Baicalin on hippocampal
neurons after status epilepticus in mice. Zhong Hua Shi Yan Wai Ke
Za Zhi She. 29:1697–1699. 2012.(In Chinese).
|
|
25
|
Gröticke I, Hoffmann K and Löscher W:
Behavioral alterations in a mouse model of temporal lobe epilepsy
induced by intrahippocampal injection of kainate. Exp Neurol.
213:71–83. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Laursen SE and Belknap JK:
Intracerebroventricular injections in mice. Some methodological
refinements. J Pharmacol Methods. 16:355–357. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Racine RJ, Steingart M and McIntyre DC:
Development of kindling-prone and kindling-resistant rats:
Selective breeding and electrophysiological studies. Epilepsy Res.
35:183–195. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pretel S, Applegate CD and Piekut D:
Apoptotic and necrotic cell death following kindling induced
seizures. Acta Histochem. 99:71–79. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cheng JY, Wu LN, Wang QZ, Gan YF, Liu GY
and Yu H: Altered mitochondria and Bcl-2 expression in the
hippocampal CA3 region in a rat model of acute epilepsy. Zhong Guo
Shen Jing Zai Sheng Yan Jiu Ying Wen Ban. 4:276–280. 2009.(In
Chinese).
|