Introduction
Alzheimer's disease (AD) is a progressive
neurodegenerative disorder which is characterized by the loss of
cognition and memory capacity, and decreased visual-spatial skills.
Pathologically, AD is characterized by increased amyloid-beta (Aβ),
hyperphosphorylation and the aggregation of tau protein (1,2). Aβ
peptides are produced from amyloid precursor protein (APP) when it
is cleaved by β- and γ- secretases into amino acid peptides within
the cerebral cortex and hippocampus (3). Accumulation of insoluble Aβ induces the
aggregation of peptide-forming amyloid fibrils, which have been
demonstrated to be neurotoxic in vitro and vivo
(4). The γ-secretase enzyme induces
intramembrane cleavage of APP, and is part of a multi-subunit
intramembranous protein complex that includes PS-1 (5,6).
Moreover, excessive deposition of Aβ stimulates
microtubule-associated protein tau aggregation into abnormally
hyperphosphorylated tau, which assemble into paired helical
filaments (PHFs) are significantly increased in patients with AD
(7).
Aβ may trigger neurodegeneration via oxidative
stress. Previous studies have demonstrated that the production of
excessive reactive oxygen species (ROS) and signs of oxidative
stress were detected in the AD brain (8–11). In
addition, it has been demonstrated that oxidative stress has a
critical role in Aβ-mediated neuronal cytotoxicity by triggering or
facilitating neurodegeneration (12). Oxidative damage may initiate during
the earlier stages of AD and induce amnestic mild cognitive
impairment and a reduced antioxidant capacity (13–15).
Furthermore, oxidative damage is associated with apoptosis,
mitochondrial membrane damage and mitochondrial dysfunction
(16); therefore, reducing oxidative
stress may decrease Aβ-induced neurotoxicity (17). We hypothesized that strong
antioxidants were capable of attenuating oxidative stress, and may
represent a therapeutic strategy to treat Aβ-induced neurotoxicity
and improve the symptoms of AD.
A previous study in transgenic animals have shown
that moderate consumption of red wine may alleviate AD-like
neuropathology and cognitive deterioration (18). Furthermore, Ono et al
(19) demonstrated that a specific
grape-derived polyphenolic extract (GSPE) reduced Aβ peptide
oligomerization and fibril development in vitro. Previous
studies have also indicated that families of polyphenols may
interfere with tau fibril formation in vitro and in cultured
cells (20,21). Notably, it has been demonstrated that
GSPE attenuates the aggregation of tau-mediated neuropathology in
the brain of the Thy-1 mutated human tau mouse model of tauopathy
(22). GSPE is a complex mixture of
proanthocyanidin monomers, oligomers and polymers, which are strong
antioxidants; therefore, the antioxidant activities of these
compounds may be beneficial to patients with AD (23,24). We
hypothesize that using monomers of aqueous grape seed
proanthocyanidin (GSPA) may induce a stronger antioxidant effect
than a complex mixture of GSPE. This may provide a new strategy for
the treatment of AD or, may at least improve the quality of life of
patients with AD.
Materials and methods
Ethics statement
All experimental protocols were approved by the
Ethics Committee of the Animal Center of Guangzhou University of
Traditional Chinese Medicine (Guangzhou, China).
Grape seed extraction
Grape seed plant material was purchased (Biovin
Naturprodukte, Ilbesheim, Germany), which was extracted for 2 h in
an ethanol:water (13:7; v/v) mixture. Following filtration of the
extracting solution, the filtrate was recovered using ethanol and
ultrafiltered. The eluent was concentrated by rapid drying with hot
gas, and the resulting eluate was lyophilized and reconstituted in
phosphate-buffered saline (PBS) at various concentrations for
biological assays and is referred to as aqueous GSPA.
High-performance liquid chromatography (HPLC) analysis
quantification of GSPA is shown in Fig.
1. Preliminary studies have demonstrated that GSPA is non-toxic
to normal C57 mice at single doses (≤7000 mg/kg) within the first
14 days (data not shown).
Reagents
Beta-amyloid peptide (Aβ25–35) and rhodamine 123
were purchased from Sigma-Aldrich (St. Louis, MO, USA). Dulbecco's
Modified Eagle Medium (DMEM) and PBS were obtained from Hyclone (GE
Healthcare Life Sciences, Logan, UT, USA). Fetal bovine serum
(FBS), horse serum (HS), 0.25% trypsin and phenol red were
purchased from Gibco (Thermo Fisher Scientific, Waltham, MA, USA).
Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide
(PI) were purchased from eBioscience, Inc., (San Diego, CA, USA).
Cell counting kit-8 (CCK-8) was purchased from Dojindo Molecular
Technologies, Inc., (Kumamoto, Japan). Penicillin and streptomycin
were purchased from Solarbio Science & Technology Co., Ltd.,
(Beijing, China). Lactate dehydrogenase (LDH) assay kit was
purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing,
China). Immobilon-P polyvinylidene difluoride membrane was
purchased from EMD Millipore (Billerica, MA, USA). Anti-APP
antibody (cat. no. ab2072) and phosphorylated (phospho) tau (cat.
no. s396) were purchased from Abcam (Cambridge, MA, USA).
Anti-β-actin (cat. no. sc-47778) was purchased from Santa Cruz
Biotechnology, Inc., (Dallas, Texas, USA). Horseradish peroxidase
(HRP)-conjugated anti-mouse IgG (cat. no. PA43002) was purchased
from Kirkegaard & Perry Laboratories, Inc., (Gaithersburg,
Maryland, USA). Donepezil hydrochloride was purchased from Eisai
China Inc., (Shanghai, China). All other reagents and chemicals
used in the present study were of analytical grade.
Animals and drug administration
A total of 30 APP/PS1 male heterozygous mice
[Swedish mutant β-amyloid precursor protein, a 9 exon presenilin 1
gene mutation; license number: SCXK (Su) (2010-0001)], aged 5–6
months and weighing 22±0.94 g, were purchased from Nanjing
University (Nanjing, China), of which 24 were APP/PS1 double
transgenic mice and remaining 6 were normal. Mice were housed
according to a 12-h light-dark cycle with ad libitum access
to food and water. Mice were randomly divided into the following
five groups: Control group [6 normal mice administered saline by
oral gavage (OG) for 2 months]; APP/PS1 model group (6 double
transgenic mice administered saline by OG for 2 months); APP/PS1
donepezil control group (6 double transgenic mice administered
donepezil hydrochloride by OG at 2 mg/kg/day for 2 months); low
dose APP/PS1-treated group (6 double transgenic mice administered
GSPA by OG at 50 mg/kg/day for 2 months); and high dose
APP/PS1-treated group (6 double transgenic mice administered GSPA
by OG at 100 mg/kg/day for 2 months).
Preparation of aggregated Aβ25–35
Aβ25–35 peptide was dissolved in deionized distilled
water at 1 mM and incubated for 7 days at 37°C to induce
aggregation, as previously described (10,25,26).
Following aggregation, the solution was stored at −20°C until
use.
Cell culture and treatment
Pheochromocytoma (PC12) cells were obtained from the
College of Pharmacy of Sun Yat-Sen University (Guangzhou, China).
Cells were seeded in 25 cm2 flasks at a density of
1×105 cells and maintained in DMEM supplemented with 100
U/ml penicillin, 100 U/ml streptomycin, 5% HS and 5% FBS at 37°C in
a humidified atmosphere of 95% air and 5% CO2. At 80%
confluence, cells were subcultured for 24 h in the same conditions
prior to incubation with various concentrations of GSPA (12.5, 25,
50 and 100 µg/ml) for 2 h. Following this, 20 µM Aβ25–35 was added
to the culture medium for an additional 24 h prior to the
initiation of the assays.
Cell viability assay
Cell viability was evaluated via quantitative
colorimetric CCK-8 and LDH assays following treatment with GSPA,
Aβ25–35 or combination therapy with both. Briefly, cells were
seeded onto 96-well culture plates at 1×104 cells/well
in DMEM and, following drug treatment, the cell cultures were
supplemented with 10 µl/well CCK-8 solution and incubated at 37°C
for 1 h. Optical density of each well was determined at 450 nm
using a microplate reader (FSA-1510; Thermo Fisher Scientific,
Inc.). Cell viability was expressed as a percentage of the
untreated controls.
For the LDH assay, 150 µl incubation medium was
collected from each well and added to an LDH assay solution. LDH
activity was measured using a microplate reader, according to the
manufacturer's instructions (Nanjing Jiancheng Bioengineering
Institute).
Assessment of apoptosis
Annexin V-FITC and PI staining was used to detect
apoptosis and necrosis following drug treatment. During the early
stages of apoptosis, membrane phosphatidylserine (PS) is
translocated from the inner lipid layer of the plasma membrane to
the outer layer in various cell types, including PC12 cells
(27). Once on the cell surface, PS
can be easily detected by staining with annexin, which is a protein
with a strong affinity to PS. Annexin was conjugated to the highly
photostable FITC. Cells in the late stage of apoptosis were
measured by conventional PI staining. The assay was directly
performed on live cells and the relative number of early and late
apoptotic cells was measured using flow cytometry. Following drug
treatment, cells (1×105 cells/plate) were washed with
PBS, harvested and subsequently centrifuged at 2,000 × g for 5 min
at 37°C. Cells were then resuspended in 1000 µl buffer and an
aliquot (190 µl) of the cell suspension was mixed with 5 µl Annexin
V-FITC and 10 µl PI and incubated for 25 min at room temperature in
the dark. Cell staining was measured with a fluorescence-activated
cell sorting (FACS) flow cytometer (BD FACSCanto II; BD
Biosciences, San Jose, CA, USA) at Ex=488 nm and Em=530 nm. Cell
apoptosis was expressed as the percentage of control cultures
incubated with Annexin V-FITC + PI, but not treated with GSPA or
Aβ25–35.
Measurement of mitochondrial membrane
potential (Ψm)
Mitochondrial Ψm was measured using rhodamine 123.
Rhodamine 123 can enter the mitochondrial matrix and induce
photoluminescent quenching that is dependent on mitochondrial
transmembrane potential (28).
Following drug treatment, PC12 cells were incubated with 5 mg/l
rhodamine 123 for 30 min at 37°C in the dark. Subsequently, cells
were washed three times with PBS and the fluorescence emission
intensity was measured with a flow cytometer at Ex=488 nm and
Em=535 nm. Intensity of fluorescence emission was expressed as the
percentage of control cells incubated in rhodamine 123 but not
treated with GSPA or Aβ25–35.
Morris water maze (MWM) test
The MWM test was used to assess alterations in the
behavior of the mice, as previously described (29). The maze was a circular pool
(diameter, 160 cm; depth, 50 cm) filled with water at 24–26°C to a
depth of 35 cm. Soluble skim milk was used to ensure the water was
opaque. A hidden platform (diameter, 8 cm) was submerged ~2 cm
below the surface of the water in the center of the designated
target quadrant. Visual cues were placed around the water maze. The
two phases of the MWM tests included an oriented navigation trial
and a spatial probe trial. Oriented navigation trials were
conducted four times/day for five days. In each trial, mice were
placed into the water in a different quadrant and given 120 sec to
find the platform and remain on the platform for 10 sec. If the
mouse failed to locate the platform within the given time, it was
guided to the platform and remained on the platform for 10 sec.
During the oriented navigation trials, behavior and the time
required for the mouse to locate the hidden platform (escape
latency) were recorded via a computerized video tracking system.
Mice that did not locate the hidden platform within the allotted
time scored a maximum of 120 sec. During the spatial probe trials,
the platform was removed and the mice were allowed to swim freely
for 120 sec. The mean search time each mouse spent in the target
quadrant was recorded to assess spatial memory ability.
Hematoxylin-eosin and Congo red
staining
All mice were sacrificed with an overdose (150
mg/kg) of pentobarbital sodium (Sigma-Aldrich, St. Louis, MO, USA).
Paraffin-embedded tissue sections were placed on slides, dewaxed
and stained with hematoxylin for 3 min, followed by eosin staining
for 3 sec. Sections were then dehydrated with alcohol, fixed with
xylene and sealed. Hippocampal histopathological abnormalities were
investigated under a light microscope. The number of cells in the
hippocampal CA1 region of each section was examined by three
independent pathologists in a blinded manner. The average number of
cells was used as the final result.
Three slides from each mouse were rehydrated in a
graded alcohol series, immersed in hematoxylin for 2 min, and
subsequently submerged in hydrochloric acid for 10 sec. Following
this, sections were rinsed in running tap water for 10 min until
they turned blue, washed twice in distilled water and stained with
Congo red for 40 min at room temperature prior to rinsing in
running tap water. Subsequently, the slides were submerged in
lithium carbonate for 5 sec and washed in running tap water.
Alcohol (80%) was used to differentiate between true staining and
nonspecific background staining. Sections were rinsed in running
tap water for 10 min, cleared in xylene, and covered with neutral
gum. Predetermined marks were used to position the light microscope
field for image collection (Olympus BX41; Olympus Corporation,
Tokyo, Japan). For blind analysis, this and all subsequent steps
were performed by an investigator unaware of the treatment
condition of each sample using an additional numbering code. Images
were collected using the ×20 objective (providing an overall
magnification of ×200) of the region of interest. For ease of
processing, all images were of the same size. The intensity of each
object was analyzed using Image J software version 1.37
software.
Western blot analysis
The right hippocampus was harvested from the mice
and stored in liquid nitrogen. Following tissue homogenization,
total proteins were extracted using the total protein extraction
reagents kit (EMD Millipore). Protein concentration was measured
using a bicinchoninic acid protein assay kit (Qian Chen
Biotechnology Company, Shanghai, China). Protein samples (20 µl)
were separated by SDS-PAGE for 70 min at 90 V and transferred to
PVDF membranes using transblotting apparatus (Bio-Rad Laboratories,
Inc., Hercules, CA, USA) for 90 min at 90 V. Membranes were blocked
with 5% (w/v) skim milk at room temperature for 30 min and
subsequently incubated at room temperature for 90 min with anti-APP
antibody (1:1,000) and phospho tau (1:2,000) primary antibodies.
The immunolabeled membranes were washed once with Tris-buffered
saline with Tween 20 (TBS-T) for 30 min followed by three separate
washes (10 min/wash). Following this, the membranes were probed
with a HRP-conjugated secondary antibody (1:2,000) at room
temperature for 1 h. To verify equal protein loading, the membranes
were incubated with monoclonal β-actin antibody (1:1,500) at room
temperature and then the same HRP-conjugated goat anti-mouse IgG
(1:2,000) at 37°C for 2 h. The membrane was washed three times with
TBS-T and the protein bands were visualized with enhanced
chemiluminescence western blotting detection reagents. The
intensity of each target protein band was analyzed using Image J
software version 1.37 (National Institutes of Health, Bethesda, MA,
USA) and expressed relative to β-actin density.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Hippocampal tissue was homogenized using an
automated homogenizer at 4°C. Total RNA was harvested from the
hippocampus, isolated using TRIzol reagent and reverse transcribed
into cDNA using RT-PCR kits according to the manufacturer's
instructions. The contents of the reaction mixture were as follows:
2 µl total RNA, 1 µl Oligo dT, 9 µl DEPC-treated water, 4 µl 5X
reaction buffer, 1 µl Ribolcok TMRNase inhibitor, 2 µl 10 mM dNTP
and 1 µl RevertAid TMV reverse transcriptase. cDNA was amplified by
RT-qPCR on an ABI Prism 7500 system (Thermo Fisher Scientific,
Inc.) using 12.5 µl 2X Maxima SYBR Green/Rox Master Mix reagent,
0.75 µl forward primer, 0.75 µl reverse primer, 2 µl template DNA
and 9 µl nuclease-free water. Expected RT-qPCR product sizes and
the primers used in this study are presented in the Table I. Samples were inactivated for 2 min
at 50°C prior to hot-start amplification. Amplification cycles were
performed as follows: 95°C for 15 min, followed by 40 cycles of
55°C for 15 sec, 60°C for 30 sec and 72°C for 30 sec. RT-qPCR was
repeated in triplicate. Data from the reactions were collected and
analyzed using ABI Prism 7500 software. Relative gene expression
levels were calculated according to the 2−ΔΔCq method
and were normalized to β-actin expression in each sample (30). Statistical analysis. Data are
expressed as means ± standard error of the mean. Multiple group
comparisons were performed using one-way analysis of variance
followed by Dunnett's test for pair-wise comparisons between
groups. SPSS 15.0 (SPSS, Inc., Chicago, IL, USA) was used for all
statistical analyses in this study. P<0.05 was considered to
indicate a statistically significant result.
 | Table I.Primers for reverse
transcription-quantitative polymerase chain reaction analysis of
β-actin and presenilin-1. |
Table I.
Primers for reverse
transcription-quantitative polymerase chain reaction analysis of
β-actin and presenilin-1.
| cDNA product | Sequence
(5′-3′) |
|---|
| β-actin | Forward,
CTGGTGAAACTCTGCGTCTG |
|
| Reverse,
AGAACAAGCGCCATACGACT |
| Presenilin-1 | Forward,
GTGGTGAAACTCTGCGTCTG |
|
| Reverse,
AGAACAAGCGCCATACGACT |
Results
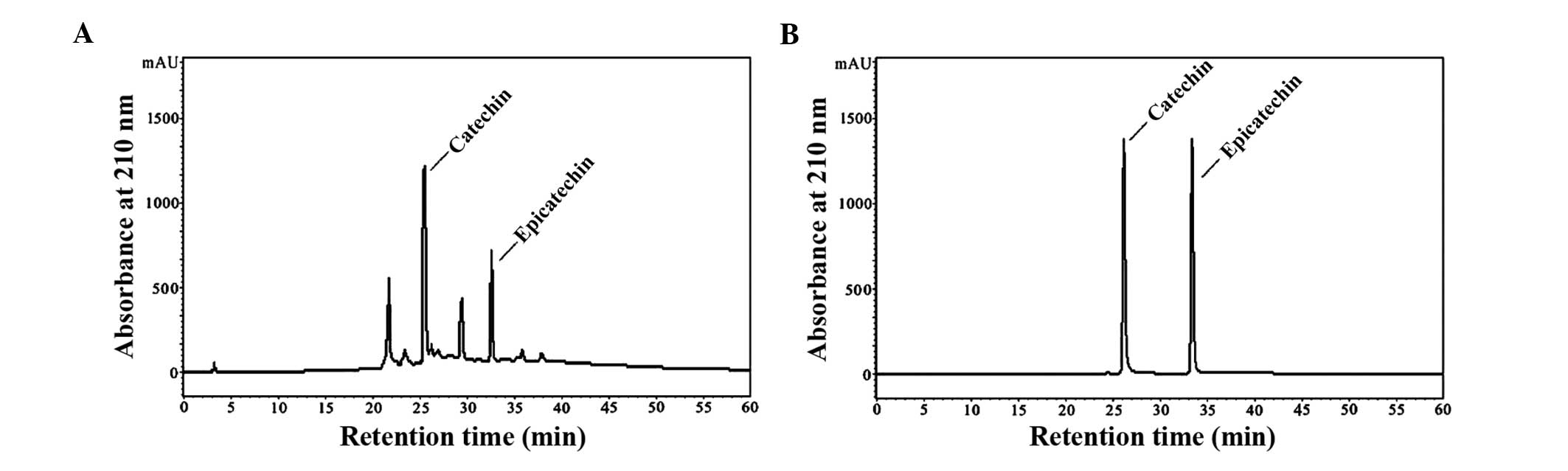
HPLC analysis of GSPA
Normal-phase-HPLC chromatograms of GSPA are
presented in Fig. 1A. The most
abundant peaks of the most active subfractions were purified using
HPLC in isocratic elution mode to yield active fractions. Catechin
retention time was 25.6 min, whereas epicatechin retention time was
32.8 min. Normal-phase-HPLC chromatograms of catechin and
epicatechin are presented in Fig.
1B. The results suggest that catechin and epicatechin are
abundant in GSPA.
Effect of GSPA on PC12 cell
viability
Cell viability was assessed using a CCK-8 reduction
assay (Fig. 2). Treatment of PC12
cells with <200 µg/ml GSPA alone for 24 h was demonstrated to be
non-toxic. CCK-8 reduction is linearly correlated with cell number
and only 200 µg/ml GSPA, which was the highest concentration
tested, reduced the viable cell number (~92% of control). The
results suggest that GSPA <200 µg/ml is non-toxic to PC12
cells.
Treatment with GSPA protects against
Aβ25–35-induced cytotoxicity in PC12 cells
Treatment of PC12 cells with 20 µM Aβ25–35 for 24 h
significantly reduced the estimated viable cell number to 65.9% of
the control (P<0.01; Fig. 3A).
However, when PC12 cells were pretreated for 2 h with GSPA (12.5,
25, 50 and 100 µg/ml, respectively) Aβ25–35-induced cytotoxicity
was significantly reduced to 69.5% of control viability at 12.5
µg/ml (P<0.05), 70.7% at 25 µg/ml (P<0.01), 84.8% at 50 µg/ml
(P<0.01), and 86% at 100 µg/ml (P<0.01).
Analysis of the levels of LDH released into the
culture media from dead/dying cells confirmed that GSPA partially
blocked Aβ25–35-induced cytotoxicity (Fig. 3B). Compared with the control group,
Aβ25–35 treatment significantly increased LDH leakage (P<0.01)
and this release was significantly attenuated by GSPA
administration (P<0.01). The results suggest that GSPA is able
to partially block Aβ25–35-induced cytotoxicity.
Treatment with GSPA reduces
Aβ25–35-induced apoptosis
Annexin V-FITC and PI double staining can
distinguish healthy cells (annexin-negative; PI-negative) from
early apoptotic (annexin-positive; PI-negative), late apoptotic
(annexin-positive; PI-positive), and necrotic (annexin-negative;
PI-positive) cells (31). The
apoptotic (early + late) rate of PC12 cells following Aβ25–35
treatment was 45.1%; however, GSPA pretreatment significantly
reduced the apoptotic rate to 39.9% at 50 µg/ml (P<0.05) and
33.9% at 100 µg/ml (P<0.01). The healthy cell rate following
Aβ25–35 treatment was 53.3%. However, GSPA pretreatment increased
the healthy cell rate to 60% at 50 µg/ml and 65.1% at 100 µg/ml
(both P<0.01, as compared with Aβ25–35 alone) (Fig. 4). The results suggest that GSPA
reduces Aβ25–35-induced apoptosis.
 | Figure 4.GSPA administration reduced
Aβ25–35-induced apoptosis. (A) Individual values (%) are listed in
representative dot plots and living PC12 cells were gated as shown
in the fluorescence-activated cell sorting histograms for control
PC12 cells, PC12 cells exposed to 20 µM Aβ25–35, 20 µM Aβ25–35 + 50
µg/ml GSPA and 20 µM Aβ25–35 + 100 µg/ml GSPA. (B) A decrease in
annexin-positive, PI-negative cells (lower right quadrant) and
annexin-positive, PI-positive cells (higher right quadrant) was
detected, which is indicative of reduced apoptosis. The increase in
annexin-negative and PI-negative cells (lower left quadrant) is
indicative of healthy non-apoptotic cells. Data are expressed as
the mean ± standard error of the mean (n=3). *P<0.01, as
compared with the control group; **P<0.01 and ***P<0.05, as
compared with the Aβ25–35-treated group. GSPA, grape seed
proanthocyanidin; Aβ25–35, amyloid β25–35 peptide, PI, propidium
iodide; FITC, fluorescein isothiocyanate. |
Treatment with GSPA reverses
depolarized mitochondrial Ψm in PC12 cells
Cells exposed to 20 µM Aβ25–35 for 24 h exhibited
significantly depolarized mitochondrial Ψm (45% loss of Ψm;
P<0.01, as compared with control group), which indicated
impending apoptosis or necrosis (Fig.
5). Pretreatment with GSPA partially reversed the response to
subsequent treatment with 20 µM Aβ25–35 for 24 h. Rhodamine 123
fluorescence percentage rate was 56% at 50 µg/ml and 88.3% at 100
µg/ml (both P<0.001, as compared with the Aβ25–35 model group).
The results suggest that GSPA is able to reverse depolarized
mitochondrial Ψm in PC12 cells.
Treatment with GSPA improves learning
ability in APP/PS1 double transgenic mice
The results of the oriented navigation trials are
presented in Table II. No
significant differences in swim speed were detected among the
groups (data not shown). The results of the MWM test demonstrated
that APP/PS1 mutant mice exhibited significantly impaired learning
ability in the water maze, as compared with the control group from
days 1–5 (P<0.05). The results of the spatial probe trials are
shown in Fig. 6. As compared with
the control group, the APP/PS model group exhibited significantly
increased escape latency times (P<0.01) and a significantly
decreased number of platform crossings (P<0.01). The APP/PS1
donepezil group exhibited significantly decreased latency
(P<0.01) and a significantly increased number of platform
crossings (P<0.01), as compared with the APP/PS1 model group.
The APP/PS1-treated low and high dose GSPA groups also exhibited
significantly reduced latency periods (P<0.05). Notably, the
high dose GPSA group exhibited an increased number of platform
crossings, as compared with the low dose group. Therefore, these
results suggested that GSPA treatment may improve the learning
ability of mice in a concentration-dependent manner. The results
suggest that GSPA is able to improve learning ability in APP/PS1
double transgenic mice.
| Table II.Improved learning ability in amyloid
precursor protein/presenilin-1 double transgenic mice treated with
grape seed proanthocyanidin (GSPA), as determined by the Morris
water maze test. |
Table II.
Improved learning ability in amyloid
precursor protein/presenilin-1 double transgenic mice treated with
grape seed proanthocyanidin (GSPA), as determined by the Morris
water maze test.
|
| Time taken to find
the platform (sec) |
|---|
|
|
|
|---|
| Group | Day 1 | Day 2 | Day 3 | Day 4 | Day 5 |
|---|
| Control | 117.50±2.50 | 75.83±20.04 | 67.17±23.70 | 58.17±20.02 | 52.67±11.11 |
| Model | 120.00±0.00 | 117.00±3.00 | 102.50±11.34 | 114.17±5.83 |
107.17±8.64a |
| Donepezil | 120.00±0.00 | 102.33±17.67 | 81.67±13.64 | 78.83±18.36 |
57.67±14.05b |
| Low GSPA | 120.00±0.00 | 97.67±7.31 | 88.33±13.18 | 81.67±10.84 |
70.17±8.66b |
| High GSPA | 120.00±0.00 | 80.00±14.09 | 82.50±13.20 |
74.50±8.72b |
67.83±3.96b |
Treatment with GSPA alleviates amyloid
plaques in the hippocampus of APP/PS1 double transgenic mice
HE staining revealed no remarkable neuronal
abnormalities in the hippocampus of mice in the control group. The
pyramidal cells in the CA1 region were arranged neatly and tightly,
the cells were round and intact with clear dark blue stained
nuclei. However, obvious hippocampal histopathological damage was
demonstrated in the APP/PS1 model group. The pyramidal layered
structure was disintegrated and neuronal loss was detected in the
CA1 region. Neurons exhibited pyknotic nuclei with a shrunken or
irregular shape. Following GSPA or donepezil hydrochloride oral
administration for 2 months these abnormalities were attenuated, as
compared with the APP/PS1 model group. Cell morphology was regular,
the cells were arranged more neatly and tightly, and cell nuclei
stained clear. The APP/PS1-treated high dose GSPA group exhibited a
stronger attenuation of these abnormalities, as compared with the
low group, suggesting a concentration-dependent effect (Fig. 7).
Congo red was used to stain the amyloid plaques in
the hippocampus of APP/PS1 double transgenic mice. The results
demonstrated that amyloid plaques were distributed in the molecular
layer of the hippocampus, and rare amyloid plaques were detected in
the pyramidal cell layer. Amyloid plaques exhibited a light red
dispersion without distinct boundaries and plaque staining was
demonstrated to be denser in the hippocampi of the APP/PS1 model
group, as compared with the hippocampi of the control group. The
molecular layer exhibited an increased percentage of positive
amyloid plaque areas, as compared with the control group. Plaque
staining of APP/PS1 in the donepezil group and the low dose and
high dose GSPA groups were lighter after GSPA or donepezil
hydrochloride oral administration for 2 months. Positively stained
areas of amyloid plaques were markedly reduced, as compared with
the APP/PS1 model group (Fig. 8).
The results suggest that GSPA is able to alleviate amyloid plaques
in the hippocampus of APP/PS1 double transgenic mice.
Treatment with GSPA decreases APP and
Tau protein expression levels in the hippocampi of APP/PS1 double
transgenic mice
Western blot analyses of APP and tau protein
expression levels are shown in Fig.
9. Low APP and tau protein expression levels were detected in
the control group. Significantly increased APP and tau protein
expression levels were detected in the hippocampi of mice in the
APP/PS1 model group (P<0.05 and P<0.01, respectively).
Treatment with oral donepezil hydrochloride for 2 months
significantly decreased APP and tau protein expression levels in
the hippocampi of the APP/PS1 donepezil group, as compared with the
APP/PS1 model group (P<0.01 and P<0.05, respectively).
Furthermore, 2-month administration of oral GSPA significantly
decreased APP and tau protein expression levels in the hippocampi
of mice in the APP/PS1-treated high dose group, as compared with
the APP/PS1 model group (P<0.05). The results suggest that GSPA
is able to decrease APP and Tau protein expression levels in the
hippocampi of APP/PS1 double transgenic mice.
Treatment with GSPA decreases PS-1
mRNA expression levels in the hippocampi of APP/PS1 double
transgenic mice
Reverse transcription-quantitative polymerase chain
reaction analysis of PS-1 mRNA expression levels was performed, as
shown in Fig. 10. PS-1 mRNA
expression levels were significantly increased in the APP/PS1 model
group, as compared with the control group (P<0.01). PS-1
expression levels in the APP/PS1 donepezil and high dose GSPA
groups were significantly decreased, as compared with the APP/PS1
model group (both P<0.01). The results suggest that GSPA is able
to decrease PS-1 mRNA expression levels in the hippocampi of
APP/PS1 double transgenic mice.
Discussion
AD is a growing public health concern with
devastating impacts. The disease is characterized by the hallmarks
of Aβ peptide accumulation with hyperphosphorylation and
aggregation of tau protein. Previous studies have indicated that
the accumulation of neurotoxic Aβ peptides has a causal role in AD
dementia and memory deficits in the Tg2576 mouse AD model (32,33). It
has also been demonstrated that certain GSPEs may interfere with
the aggregations of synthetic Aβ peptides in vitro (34). Furthermore, according to the results
of a previous binding assay, GSPE is capable of inhibiting tau
peptide polymerization and scattering pre-aggregated tau peptide
(35).
Oxidative stress has an important role in the
pathophysiology of AD. Previous studies have indicated that
oxidative stress leads to apoptosis and excessive ROS facilitates
neuronal apoptosis in Aβ-induced neuronal cell death (36,37).
Moreover, it has been demonstrated that the overproduction of Aβ
leads to Aβ-associated free-radical production and cell death
(37,38). These findings indicate that Aβ
induces oxidative stress and vice versa, which leads to more
oxidative damage, including membrane damage and reduced cell
viability. When cell membranes are damaged, LDH levels increase.
The present study demonstrated that GSPA is able to reverse
Aβ25–35-induced LDH leakage and decreased PC12 cell viability
(39). These results demonstrated
that GSPA is capable of scavenging oxygen radicals. Therefore, the
protective effect of GSPA against Aβ25–35 induced oxidative stress
in PC12 cells may be due to enhanced anti-oxidant capacity and
reduced accumulation of intracellular ROS.
Cell apoptosis has a central role in the
pathogenesis of AD. Oxidative stress induces apoptotic cell death
via the activation of caspase 3 (40). In the present study, Annexin V-FITC
and PI double staining was used to investigate basal apoptosis
rates. PC12 cells treated with extracellular Aβ25–35 at
concentrations of 20 µM exhibited significantly increased levels of
apoptosis, as compared with the control cells. These data suggested
that Aβ25–35 may increase caspase 3 expression levels and induce
oxidative stress, leading to apoptosis. However, treatment with 50
or 100 µg/ml GSPA significantly decreased cell apoptosis. This
result may be associated with decreasing levels of caspase 3 and
oxidative stress.
A significant reduction of the mitochondrial Ψm can
trigger apoptosis through the release of caspase 3. Previous
studies have demonstrated that Aβ25–35 leads to oxidative stress
and mitochondrial damage due to the loss of mitochondrial Ψm
(41,42). Consistent with a previous study
(43), the results of the present
study demonstrated a significant decrease in mitochondrial Ψm
following stimulation with extracellular Aβ25–35, as compared with
the control cells, using rhodamine 123 fluorescence. Pretreatment
of PC12 cells with GSPA reversed the mitochondrial Ψm
depolarization induced by Aβ25–35, which suggested that GSPA may
prevent cell death by blocking the activation of the mitochondrial
apoptosis pathway.
In the present study, the significant cytotoxic
effect of Aβ25–35 on PC12 cells was demonstrated by the CCK-8
assay, and Annexin V-FITC and PI double staining with flow
cytometry. It was hypothesized that activation of caspase 3 induced
apoptosis. This result may be associated with oxidative stress and
stress-related damage to the mitochondria or its membranes.
Notably, GSPA may reverse the Aβ25–35-induced increase of ROS and
decrease of mitochondrial Ψm, which ultimately protected the cells
from apoptosis.
The results of the present study also demonstrated
that oral administration of GSPA significantly reduced the
accumulation of Aβ peptides, alleviated the hyperphosphorylation of
tau protein and improved learning behavior and memory in APP/PS1
double transgenic mice. The mechanisms underlying these phenomenon
may involve the downregulation of Aβ accumulation and the
disruption of tau protein hyperphosphorylation in the brain.
APP/PS1 mice exhibited a significant loss of
learning, cognition and memory behavior in the present study, as
compared with the control mice. These results are consistent with
previous studies, which have demonstrated APP/PS1 mice with early
onset brain amyloidosis, and synaptic changes in the hippocampus
and brain cortex (44,45). The MWM test is a well-known
behavioral task which is used to assess oriented navigation and
spatial probe memory capacity (46).
During the spatial probe phase of the MWM test in the present
study, the GSPA treatment group exhibited significant decreased
escape latency periods and an increased number of platform crossing
incidences, as compared with APP/PS1 model group. These data
demonstrated that the GSPA-treated APP/PS1 mice exhibited an
enhanced capacity for learning, cognition and memory. We
hypothesize that this effect may be induced by a reduction in Aβ in
the brain, as excessive toxic Aβ has been demonstrated to induce
the formation of Aβ plaques (47).
HE and Congo red staining was used to detect amyloidosis in the
hippocampi of the mice. The APP/PS1 model group exhibited a
significant increase in the number of Aβ plaques in the
hippocampus, as compared with the control group. Notably, GSPA
treatment significantly ameliorated these Aβ plaques.
The results of the present study have demonstrated
that mutations in the APP and PS genes increase oxidative stress in
the neurons of APP/PS1 mice, and oxidative stress is mediated by
Aβ. The Aβ protein is critical to the pathogenesis of AD (48,49);
however, the precise mechanism underlying the pathogenesis of AD is
yet to be fully elucidated. Therefore, one of the aims of
preventive or early treatment of AD is to decrease oxidative
stress. Aβ peptides are produced from the APP, which are cleaved by
β- and γ- secretases into amino acid peptides within the cerebral
cortex and hippocampus (3).
γ-secretase is an intramembrane aspartyl protease that is
critically involved in AD via the proteolysis of APP, which
generates the pathogenic and amyloid plaque-forming Aβ1–42 peptide
(50). PS-1 is a member of a
multi-subunit intramembranous protein complex with γ-secretase;
thus, a reduction in PS-1 mRNA expression levels directly
suppresses the activation of γ-secretase. PS-1 also stimulates tau
protein aggregation into abnormally hyperphosphorylated tau via Aβ
(51). Therefore, it was
hypothesized that restraining PS-1 mRNA and APP protein expression
levels via decreased oxidative stress may be a viable strategy for
AD therapy. In the present study, western blot and RT-qPCR analyses
were used to confirm that treatment with GSPA significantly
decreased PS-1 mRNA expression levels. The effects of GSPA on PS-1
mRNA expression and γ-secretase activation may interrupt the
generation of the Aβ peptide. Moreover, the results of the present
study demonstrated that oral treatment with GSPA significantly
reduced the accumulation of APP and insoluble tau in the brains of
APP/PS1 mice. GSPA may function as a strong antioxidant by
decreasing the levels of APP and tau protein. This phenomenon may
be associated with: i) the strong antioxidant action of GSPA
suppressing the activation of γ- secretases to reduce Aβ
production; or ii) GSPA directly interfering with the aggregation
of Aβ or dissociated aggregation of Aβ into oligomers, which may
reduce oxidative stress and restore the antioxidant balance.
In conclusion, the present preclinical study
demonstrated that GSPA treatment attenuates APP production or
accumulation, interrupts the aggregation of neurotoxic Aβ and
decreases hyperphosphorylated tau deposition. Notably, GSPA
treatment improved the cognition and memory capacity of a APP/PS1
double transgenic mouse model. The results of the present in
vitro biochemistry studies and in vivo preclinical
studies suggested that GSPA administration may benefit AD via two,
non-exclusive mechanisms: i) enhanced antioxidant function and ii)
downregulation of caspase 3-mediated aggregation of Aβ in response
to oxidative stress. The results of the present study provide
support for continuing the development of GSPA for the treatment
and/or prevention of AD.
Acknowledgements
The present study was supported by the Special Funds
from Central Finance of China in Support of the Development of
Local Colleges and University [grant no. 276 (2014)]. This research
was also supported in part by BannerBio Nutraceuticals, Inc. The
funding bodies had no role in study design, data collection and
analysis, decision to publish, or preparation of the
manuscript.
References
|
1
|
Blennow K, de Leon MJ and Zetterberg H:
Alzheimer's disease. Lancet. 368:387–403. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nelson PT, Braak H and Markesbery WR:
Neuropathology and cognitive impairment in Alzheimer disease: A
complex but coherent relationship. J Neuropathol Exp Neurol.
68:1–14. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Haass C and Selkoe DJ: Soluble protein
oligomers in neurodegeneration: Lessons from the Alzheimer's
amyloid beta-peptide. Nat Rev Mol Cell Biol. 8:101–112. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li MH, Jang JH, Sun B and Surh YJ:
Protective effects of oligomers of grape seed polyphenols against
beta-amyloid-induced oxidative cell death. Ann. NY Acad Sci.
1030:317–329. 2004. View Article : Google Scholar
|
|
5
|
Frykman S, Hur JY, Frånberg J, Aoki M,
Winblad B, Nahalkova J, Behbahani H and Tjernberg LO: Synaptic and
endosomal localization of active γ-secretase in rat brain. PLoS
One. 5:e89482010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Smolarkiewicz M, Skrzypczak T and
Wojtaszek P: The very many faces of presenilins and the γ-secretase
complex. Protoplasma. 250:997–1011. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hasegawa M, Morishima-Kawashima M, Takio
K, Suzuki M, Titani K and Ihara Y: Protein sequence and mass
spectrometric analyses of tau in the Alzheimer's disease brain. J
Biol Chem. 267:17047–17054. 1992.PubMed/NCBI
|
|
8
|
Crack PJ and Taylor JM: Reactive oxygen
species and the modulation of stroke. Free Radic Biol Med.
38:1433–1444. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hu JF, Chu SF, Ning N, Yuan YH, Xue W,
Chen NH and Zhang JT: Protective effect of (−)clausenamide against
Abeta-induced neurotoxicity in differentiated PC12 cells. Neurosci
Lett. 483:78–82. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li G, Ma R, Huang C, Tang Q, Fu Q, Liu H,
Hu B and Xiang J: Protective effect of erythropoietin on
beta-amyloid-induced PC12 cell death through antioxidant
mechanisms. Neurosci Lett. 442:143–147. 2008a. View Article : Google Scholar
|
|
11
|
Zhang HY, Liu YH, Wang HQ, Xu JH and Hu
HT: Puerarin protects PC12 cells against beta-amyloid-induced cell
injury. Cell Biol Int. 32:1230–1237. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Crack PJ, Cimdins K, Ali U, Hertzog PJ and
Iannello RC: Lack of glutathione peroxidase-1 exacerbates
Abeta-mediated neurotoxicity in cortical neurons. J Neural Transm.
113:645–657. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Petersen RC: Mild cognitive impairment:
Transition between aging and Alzheimer's disease. Neurologia.
15:93–101. 2000.PubMed/NCBI
|
|
14
|
Rinaldi P, Polidori MC, Metastasio A,
Mariani E, Mattioli P, Cherubini A, Catani M, Cecchetti R, Senin U
and Mecocci P: Plasma antioxidants are similarly depleted in mild
cognitive impairment and in Alzheimer's disease. Neurobiol Aging.
24:915–919. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Guidi I, Galimberti D, Lonati S,
Novembrino C, Bamonti F, Tiriticco M, Fenoglio C, Venturelli E,
Baron P, Bresolin N and Scarpini E: Oxidative imbalance in patients
with mild cognitive impairment and Alzheimer's disease. Neurobiol
Aging. 27:262–269. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen JX and Yan SD: Pathogenic role of
mitochondrial amyloid- beta peptide. Expert Rev Neurother.
7:1517–1525. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee SY, Lee JW, Lee H, Yoo HS, Yun YP, Oh
KW, Ha TY and Hong JT: Inhibitory effect of green tea extract on
beta-amyloid-induced PC12 cell death by inhibition of the
activation of NF-kappaB and ERK/p38 MAP kinase pathway through
antioxidant mechanisms. Brain Res Mol Brain Res. 140:45–54. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang J, Ho L, Zhao Z, Seror I, Humala N,
Dickstein DL, Thiyagarajan M, Percival SS, Talcott ST and Pasinetti
GM: Moderate consumption of Cabernet Sauvignon attenuates Abeta
neuropathology in a mouse model of Alzheimer's disease. FASEB J.
20:2313–2320. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ono K, Condron MM, Ho L, Wang J, Zhao W,
Pasinetti GM and Teplow DB: Effects of grape seed-derived
polyphenols on amyloid beta-protein self-assembly and cytotoxicity.
J Biol Chem. 283:32176–32187. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Crowe A, Ballatore C, Hyde E, Trojanowski
JQ and Lee VM: High throughput screening for small molecule
inhibitors of heparin-induced tau fibril formation. Biochem Biophys
Res Commun. 358:1–6. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pickhardt M, von Bergen M, Gazova Z,
Hascher A, Biernat J, Mandelkow EM and Mandelkow E: Screening for
inhibitors of tau polymerization. Curr Alzheimer Res. 2:219–226.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang J, Santa-Maria I, Ho L,
Ksiezak-Reding H, Ono K, Teplow DB and Pasinetti GM: Grape derived
polyphenols attenuate tau neuropathology in a mouse model of
Alzheimer's disease. J Alzheimers Dis. 22:653–661. 2010.PubMed/NCBI
|
|
23
|
Okello EJ, Savelev SU and Perry EK: In
vitro anti-beta-secretase and dual anti-cholinesterase activities
of Camellia sinensis L. (tea) relevant to treatment of
dementia. Phytother Res. 18:624–627. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang J, Ho L, Zhao W, Ono K, Rosensweig C,
Chen L, Humala N, Teplow DB and Pasinetti GM: Grape-derived
polyphenolics prevent Abeta oligomerization and attenuate cognitive
deterioration in a mouse model of Alzheimer's disease. J Neurosci.
28:6388–6392. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li J, Zhang HL, Wang Z, Liang YM, Jiang L,
Ma W and Yang DP: Determination content of the antidepressant
extraction and analysis the trace elements from Morinda
officinalis. Zhong Yao Cai. 31:1337–1340. 2008b.(In
Chinese).
|
|
26
|
Labbé JF, Lefèvre T, Guay-Bégin AA and
Auger M: Structure and membrane interactions of the β-amyloid
fragment 25–35 as viewed using spectroscopic approaches. Phys Chem
Chem Phys. 15:7228–7239. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Emoto K, Toyama-Sorimachi N, Karasuyama H,
Inoue K and Umeda M: Exposure of phosphatidylethanolamine on the
surface of apoptotic cells. Exp Cell Res. 232:430–434. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Scaduto RC Jr and Grotyohann LW:
Measurement of mitochondrial membrane potential using fluorescent
rhodamine derivatives. Biophys J. 76:469–477. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee MR, Yun BS, Park SY, Ly SY, Kim SN,
Han BH and Sung CK: Anti-amnesic effect of Chong-Myung-Tang on
scopolamine-induced memory impairments in mice. J Ethnopharmacol.
132:70–74. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCt method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen S, Cheng AC, Wang MS and Peng X:
Detection of apoptosis induced by new type gosling viral enteritis
virus in vitro through fluorescein annexin V-FITC/PI double
labeling. World J Gastroenterol. 14:2174–2178. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cleary JP, Walsh DM, Hofmeister JJ,
Shankar GM, Kuskowski MA, Selkoe DJ and Ashe KH: Natural oligomers
of the amyloid-beta protein specifically disrupt cognitive
function. Nat Neurosci. 8:79–84. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Klyubin I, Walsh DM, Lemere CA, Cullen WK,
Shankar GM, Betts V, Spooner ET, Jiang L, Anwyl R, Selkoe DJ and
Rowan MJ: Amyloid beta protein immunotherapy neutralizes Abeta
oligomers that disrupt synaptic plasticity in vivo. Nat Med.
11:556–561. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Porat Y, Abramowitz A and Gazit E:
Inhibition of amyloid fibril formation by polyphenols: Structural
similarity and aromatic interactions as a common inhibition
mechanism. Chem Biol Drug Des. 67:27–37. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ho L, Yemul S, Wang J and Pasinetti GM:
Grape seed polyphenolic extract as a potential novel therapeutic
agent in tauopathies. J Alzheimers Dis. 16:433–439. 2009.PubMed/NCBI
|
|
36
|
Bonda DJ, Wang X, Perry G, Nunomura A,
Tabaton M, Zhu X and Smith MA: Oxidative stress in Alzheimer
disease: A possibility for prevention. Neuropharmacology.
59:290–294. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kadowaki H, Nishitoh H, Urano F, Sadamitsu
C, Matsuzawa A, Takeda K, Masutani H, Yodoi J, Urano Y, Nagano T
and Ichijo H: Amyloid beta induces neuronal cell death through
ROS-mediated ASK1 activation. Cell Death Differ. 12:19–24. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sponne I, Fifre A, Drouet B, Klein C,
Koziel V, Pinçon-Raymond M, Olivier JL, Chambaz J and Pillot T:
Apoptotic neuronal cell death induced by the non-fibrillar
amyloid-beta peptide proceeds through an early reactive oxygen
species-dependent cytoskeleton perturbation. J Biol Chem.
278:3437–3445. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang YM: Protective effect of quercetin
on aroclor 1254-induced oxidative damage in cultured chicken
spermatogonial cells. Toxicol Sci. 88:545–550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Marques CA, Keil U, Bonert A, Steiner B,
Haass C, Muller WE and Eckert A: Neurotoxic mechanisms caused by
the Alzheimer's disease-linked Swedish amyloid precursor protein
mutation: Oxidative stress, caspases, and the JNK pathway. J Biol
Chem. 278:28294–28302. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yankner BA, Dawes LR, Fisher S,
Villa-Komaroff L, Oster-Granite ML and Neve RL: Neurotoxicity of a
fragment of the amyloid precursor associated with Alzheimer's
disease. Science. 245:417–420. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jang JH and Surh YJ: Protective effect of
resveratrol on beta-amyloid-induced oxidative PC12 cell death. Free
Radic Biol Med. 34:1100–1110. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hoxha E, Boda E, Montarolo F, Parolisi R
and Tempia F: Excitability and synaptic alterations in the
cerebellum of APP/PS1 mice. PLoS One. 7:e347262012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Park SM, Shin JH, Moon GJ, Cho SI, Lee YB
and Gwag BJ: Effects of collagen-induced rheumatoid arthritis on
amyloidosis and microvascular pathology in APP/PS1 mice. BMC
Neurosci. 12:1062011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sharma S, Rakoczy S and Brown-Borg H:
Assessment of spatial memory in mice. Life Sci. 87:521–536. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Galimberti D and Scarpini E: Alzheimer's
disease: From pathogenesis to disease-modifying approaches. CNS
Neurol Disord Drug Targets. 10:163–174. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lovell MA, Xie C, Xiong S and Markesbery
WR: Protection against amyloid beta peptide and iron/hydrogen
peroxide toxicity by alpha lipoic acid. J Alzheimers Dis.
5:229–239. 2003.PubMed/NCBI
|
|
49
|
Butterfield DA, Castegna A, Lauderback CM
and Drake J: Evidence that amyloid beta-peptide-induced lipid
peroxidation and its sequelae in Alzheimer's disease brain
contribute to neuronal death. Neurobiol Aging. 23:655–664. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hass MR, Sato C, Kopan R and Zhao G:
Presenilin: RIP and beyond. Semin Cell Dev Biol. 20:201–210. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kong R, Chang S, Xia W and Wong ST:
Molecular dynamics simulation study reveals potential substrate
entry path into γ-secretase/presenilin-1. J Struct Biol.
191:120–129. 2015. View Article : Google Scholar : PubMed/NCBI
|